




–°—З–Є—В–∞–µ—В—Б—П, —З—В–Њ —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ –±–Њ–ї–µ–µ 80 –≥ —Н—В–∞–љ–Њ–ї–∞ –≤ —Б—Г—В–Ї–Є (1 –ї –≤–Є–љ–∞, –Њ–Ї–Њ–ї–Њ 3,6 –ї –њ–Є–≤–∞ –Є –њ—А–Є–±–ї–Є–Ј–Є—В–µ–ї—М–љ–Њ 250 –≥ –Ї—А–µ–њ–Ї–Є—Е —Б–њ–Є—А—В–љ—Л—Е –љ–∞–њ–Є—В–Ї–Њ–≤) –Љ–Њ–ґ–µ—В –њ—А–Є–≤–µ—Б—В–Є –Ї –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є —П–≤–љ–Њ–Љ—Г –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—О –њ–µ—З–µ–љ–Є —Г –Љ—Г–ґ—З–Є–љ. –£ –ґ–µ–љ—Й–Є–љ —Н—В–∞ –і–Њ–Ј–∞ –≤ 2–4 —А–∞–Ј–∞ –Љ–µ–љ—М—И–µ. –Ю–і–љ–∞–Ї–Њ —А–Є—Б–Ї —А–∞–Ј–≤–Є—В–Є—П –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є —Б–Њ–њ—А—П–ґ–µ–љ —Б –≥–Њ—А–∞–Ј–і–Њ –Љ–µ–љ—М—И–µ–є –і–Њ–Ј–Њ–є, –∞ –Є–Љ–µ–љ–љ–Њ —Б —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ–Љ 30 –≥ —Н—В–∞–љ–Њ–ї–∞ –≤ —Б—Г—В–Ї–Є [1].
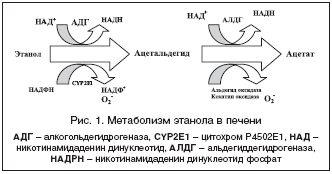
–Ь–µ—В–∞–±–Њ–ї–Є–Ј–Љ —Н—В–∞–љ–Њ–ї–∞ –≤ –њ–µ—З–µ–љ–Є. –Т –Њ–Ї–Є—Б–ї–µ–љ–Є–Є —Н—В–∞–љ–Њ–ї–∞ –≤ –њ–µ—З–µ–љ–Є –њ—А–Є–љ–Є–Љ–∞—О—В —Г—З–∞—Б—В–Є–µ —В—А–Є —Д–µ—А–Љ–µ–љ—В–љ—Л–µ —Б–Є—Б—В–µ–Љ—Л: –∞–ї–Ї–Њ–≥–Њ–ї—М–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј—Л, —Ж–Є—В–Њ—Е—А–Њ–Љ P4502E1 –Є –Ї–∞—В–∞–ї–∞–Ј—Л. –Ъ–∞—В–∞–ї–∞–Ј–љ–∞—П —Б–Є—Б—В–µ–Љ–∞ –Є–≥—А–∞–µ—В –Љ–Є–љ–Є–Љ–∞–ї—М–љ—Г—О —А–Њ–ї—М –≤ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–µ —Н—В–∞–љ–Њ–ї–∞. –Р–ї–Ї–Њ–≥–Њ–ї—М–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј—Л – —Н—В–Њ —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є–µ —Д–µ—А–Љ–µ–љ—В—Л, —Б—Г—Й–µ—Б—В–≤—Г—О—Й–Є–µ –≤ –≤–Є–і–µ –Є–Ј–Њ—Д–Њ—А–Љ –≤ –њ–µ—З–µ–љ–Є —З–µ–ї–Њ–≤–µ–Ї–∞. –Ю–љ–Є –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л –Ј–∞ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ –∞–ї–Ї–Њ–≥–Њ–ї—П, –Ї–Њ–≥–і–∞ –µ–≥–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –≤ –Ї—А–Њ–≤–Є –Є —В–Ї–∞–љ—П—Е –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–∞ –љ–Є–Ј–Ї–∞. –Я—А–Є —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–Є –±–Њ–ї—М—И–Є—Е –Ї–Њ–ї–Є—З–µ—Б—В–≤ –∞–ї–Ї–Њ–≥–Њ–ї—П –Є –њ—А–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–Љ –њ—А–Є–µ–Љ–µ —Н—В–∞–љ–Њ–ї –∞–Ї—В–Є–≤–Є—А—Г–µ—В —Б–Є—Б—В–µ–Љ—Г —Ж–Є—В–Њ—Е—А–Њ–Љ–∞ (CYP2E1). –Ш –∞–ї–Ї–Њ–≥–Њ–ї—М–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–∞, –Є –°YP2E1 –њ—А–µ–≤—А–∞—Й–∞—О—В —Н—В–∞–љ–Њ–ї –≤ –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і. –Ч–∞—В–µ–Љ –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і –њ—А–µ–≤—А–∞—Й–∞–µ—В—Б—П –∞–ї—М–і–µ–≥–Є–і–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–Њ–є –≤ –∞—Ж–µ—В–∞—В. –†–µ–і–Ї–Њ –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Є—А—Г–µ—В—Б—П –≤ –∞—Ж–µ—В–∞—В –і—А—Г–≥–Є–Љ–Є —Д–µ—А–Љ–µ–љ—В–∞–Љ–Є: –∞–ї—М–і–µ–≥–Є–і–Њ–Ї—Б–Є–і–∞–Ј–Њ–є –Є –Ї—Б–∞–љ—В–Є–љ–Њ–Ї—Б–Є–і–∞–Ј–Њ–є. –Р—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і – –≤—Л—Б–Њ–Ї–Њ—А–µ–∞–Ї—В–Є–≤–љ—Л–є –Є –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ–Њ —В–Њ–Ї—Б–Є—З–љ—Л–є –Љ–µ—В–∞–±–Њ–ї–Є—В —Н—В–∞–љ–Њ–ї–∞. –Ъ —Б—З–∞—Б—В—М—О, –њ–µ—З–µ–љ—М —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –±—Л—Б—В—А–Њ–є –Є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ–є —Н–ї–Є–Љ–Є–љ–∞—Ж–Є–Є —Н—В–Њ–≥–Њ —Б–Њ–µ–і–Є–љ–µ–љ–Є—П. –Х—Б–ї–Є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –њ–µ—З–µ–љ–Є —Г–і–∞–ї—П—В—М –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і –љ–∞—А—Г—И–∞–µ—В—Б—П, —В–Њ –Њ–љ –љ–∞–Ї–∞–њ–ї–Є–≤–∞–µ—В—Б—П –≤ –њ–µ—З–µ–љ–Є –Є –Ї—А–Њ–≤–Є. –Ш–Љ–µ–љ–љ–Њ –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і –≤—Л–Ј—Л–≤–∞–µ—В –њ–Њ–Ї—А–∞—Б–љ–µ–љ–Є–µ –ї–Є—Ж–∞, —В–∞—Е–Є–Ї–∞—А–і–Є—О, –∞ –Є–љ–Њ–≥–і–∞ –Є —Б–Њ—Б—Г–і–Є—Б—В—Л–є –Ї–Њ–ї–ї–∞–њ—Б (—А–Є—Б. 1) [2,3].
–Ь–µ—В–∞–±–Њ–ї–Є–Ј–Љ —Н—В–∞–љ–Њ–ї–∞ –≤ –ґ–µ–ї—Г–і–Ї–µ. –•–Њ—В—П –њ–µ—З–µ–љ—М – –Њ—Б–љ–Њ–≤–љ–Њ–µ –Љ–µ—Б—В–Њ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ —Н—В–∞–љ–Њ–ї–∞, –љ–Њ –Њ–љ —В–∞–Ї–ґ–µ –Њ–Ї–Є—Б–ї—П–µ—В—Б—П –Є –≤ –ґ–µ–ї—Г–і–Њ—З–љ–Њ––Ї–Є—И–µ—З–љ–Њ–Љ —В—А–∞–Ї—В–µ. –Ш–Ј–Њ—Д–Њ—А–Љ—Л –∞–ї–Ї–Њ–≥–Њ–ї—М–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј—Л –±—Л–ї–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –≤ –ґ–µ–ї—Г–і–Ї–µ –Є –Ї–Є—И–µ—З–љ–Є–Ї–µ. –Ц–µ–ї—Г–і–Њ—З–љ–∞—П –∞–ї–Ї–Њ–≥–Њ–ї—М–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–∞ –Њ—Б—Г—Й–µ—Б—В–≤–ї—П–µ—В –њ–µ—А–≤—Л–є —Н—В–∞–њ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ —Н—В–∞–љ–Њ–ї–∞, –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ –Ї–Њ—В–Њ—А–Њ–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ —Н—В–∞–љ–Њ–ї–∞, –њ–Њ—Б—В—Г–њ–∞—О—Й–µ–≥–Њ –≤ –њ–Њ—А—В–∞–ї—М–љ—Л–є –Ї—А–Њ–≤–Њ—В–Њ–Ї, —Г–Љ–µ–љ—М—И–∞–µ—В—Б—П. –Р–Ї—В–Є–≤–љ–Њ—Б—В—М –ґ–µ–ї—Г–і–Њ—З–љ–Њ–є –∞–ї–Ї–Њ–≥–Њ–ї—М–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј—Л –Љ–µ–љ—М—И–µ —Г –ґ–µ–љ—Й–Є–љ –Є –Љ–Њ–ґ–µ—В –њ–Њ–і–∞–≤–ї—П—В—М—Б—П —А—П–і–Њ–Љ –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ (–љ–∞–њ—А–Є–Љ–µ—А, –∞—Ж–µ—В–Є–ї—Б–∞–ї–Є—Ж–Є–ї–Њ–≤–Њ–є –Ї–Є—Б–ї–Њ—В–Њ–є –Є–ї–Є –±–ї–Њ–Ї–∞—В–Њ—А–∞–Љ–Є –≥–Є—Б—В–∞–Љ–Є–љ–Њ–≤—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ II —В–Є–њ–∞) [2,3].
–Я–∞—В–Њ–≥–µ–љ–µ–Ј –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є. –Ю–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ–µ –∞–ї–Ї–Њ–≥–Њ–ї—М–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј–Њ–є –Њ–Ї–Є—Б–ї–µ–љ–Є–µ —Н—В–∞–љ–Њ–ї–∞ —Б–≤—П–Ј–∞–љ–Њ —Б –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є–µ–Љ –Њ–Ї–Є—Б–ї–µ–љ–љ–Њ–≥–Њ –љ–Є–Ї–Њ—В–Є–љ–∞–Љ–Є–і–∞–і–µ–љ–Є–љ –і–Є–љ—Г–Ї–ї–µ–Њ—В–Є–і–∞ (–Э–Р–Ф+) –і–Њ –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–љ–Њ–≥–Њ –љ–Є–Ї–Њ—В–Є–љ–∞–Љ–Є–і–∞–і–µ–љ–Є–љ –і–Є–љ—Г–Ї–ї–µ–Њ—В–Є–і–∞ (–Э–Р–Ф–Э) (—А–Є—Б. 1). –Ш–Ј–±—Л—В–Њ—З–љ–Њ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –Э–Р–Ф–Э –њ—А–Є–≤–Њ–і–Є—В –Ї —Б–і–≤–Є–≥—Г –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ–Њ––≤–Њ—Б—Б—В–∞–љ–Њ–≤–Є—В–µ–ї—М–љ–Њ–≥–Њ –њ–Њ—В–µ–љ—Ж–Є–∞–ї–∞ –≥–µ–њ–∞—В–Њ—Ж–Є—В–∞ –Є –Є–Ј–Љ–µ–љ–µ–љ–Є—О –і—А—Г–≥–Є—Е –Э–Р–Ф+––Ј–∞–≤–Є—Б–Є–Љ—Л—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤, –≤–Ї–ї—О—З–∞—П –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ –ї–Є–њ–Є–і–Њ–≤ –Є —Г–≥–ї–µ–≤–Њ–і–Њ–≤. –Я–Њ—Б–ї–µ–і—Б—В–≤–Є—П —Н—В–Є—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ – —Б—В–Є–Љ—Г–ї—П—Ж–Є—П —Б–Є–љ—В–µ–Ј–∞ –ґ–Є—А–љ—Л—Е –Ї–Є—Б–ї–Њ—В, –њ–Њ–і–∞–≤–ї–µ–љ–Є–µ β––Њ–Ї–Є—Б–ї–µ–љ–Є—П –≤ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є—П—Е, –љ–∞—А—Г—И–µ–љ–Є–µ –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ –≥–ї—О–Ї–Њ–љ–µ–Њ–≥–µ–љ–µ–Ј–∞ –Ј–∞ —Б—З–µ—В —Г–Љ–µ–љ—М—И–µ–љ–Є—П –µ–≥–Њ —Б—Г–±—Б—В—А–∞—В–Њ–≤, –≤ —З–∞—Б—В–љ–Њ—Б—В–Є, –Њ–Ї—Б–∞–ї–Њ–∞—Ж–µ—В–∞—В–∞, –њ–Є—А—Г–≤–∞—В–∞ –Є –і–Є–≥–Є–і—А–Њ–Ї—Б–Є–∞—Ж–µ—В–∞—В—Д–Њ—Б—Д–∞—В–∞. –Т—Б–µ —Н—В–Њ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—О –ґ–Є—А–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤ —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–µ –≥–µ–њ–∞—В–Њ—Ж–Є—В–∞, –≥–і–µ –Њ–љ–Є —Н—Б—В–µ—А–Є—Д–Є—Ж–Є—А—Г—О—В—Б—П –Є –Њ—В–Ї–ї–∞–і—Л–≤–∞—О—В—Б—П –≤ –≤–Є–і–µ —В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–Њ–≤, –њ—А–Є–≤–Њ–і—П, –≤ –Ї–Њ–љ–µ—З–љ–Њ–Љ –Є—В–Њ–≥–µ, –Ї —А–∞–Ј–≤–Є—В–Є—О —Б—В–µ–∞—В–Њ–Ј–∞ –≤ –њ–µ—З–µ–љ–Є. –Я—А–Є –љ–µ–і–Њ—Б—В–∞—В–Ї–µ —Г–≥–ї–µ–≤–Њ–і–Њ–≤ –≤ —А–∞—Ж–Є–Њ–љ–µ –њ–Є—В–∞–љ–Є—П –љ–∞—А—Г—И–µ–љ–Є–µ –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ –≥–ї—О–Ї–Њ–љ–µ–Њ–≥–µ–љ–µ–Ј–∞ –Њ–±—Г—Б–ї–Њ–≤–ї–Є–≤–∞–µ—В –≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є–µ —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ –≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є–Є [3,4].
–°–і–≤–Є–≥ –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ–Њ––≤–Њ—Б—Б—В–∞–љ–Њ–≤–Є—В–µ–ї—М–љ–Њ–≥–Њ –њ–Њ—В–µ–љ—Ж–Є–∞–ї–∞ –≤ —Б—В–Њ—А–Њ–љ—Г –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –Є –њ–Њ—Б–ї–µ–і—Г—О—Й–Є–µ —А–µ–∞–Ї—Ж–Є–Є, –Њ–њ–Є—Б–∞–љ–љ—Л–µ –≤—Л—И–µ, —Е–∞—А–∞–Ї—В–µ—А–љ—Л –і–ї—П –Њ—Б—В—А—Л—Е –њ–Њ—Б–ї–µ–і—Б—В–≤–Є–є –Њ–Ї–Є—Б–ї–µ–љ–Є—П —Н—В–∞–љ–Њ–ї–∞. –Ю–љ–Є –Њ–±—А–∞—В–Є–Љ—Л –≤ —Б–ї—Г—З–∞–µ –∞–±—Б—В–Є–љ–µ–љ—Ж–Є–Є. –Я—А–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–Љ —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–Є –∞–ї–Ї–Њ–≥–Њ–ї—П –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –≥–µ–њ–∞—В–Њ—Ж–Є—В–∞ –Є –љ–∞—А—Г—И–µ–љ–Є–µ –њ–Њ–≤—В–Њ—А–љ–Њ–≥–Њ –Њ–Ї–Є—Б–ї–µ–љ–Є—П –Э–Р–Ф–Э –≤ –Э–Р–Ф+.
–Ю–Ї–Є—Б–ї–µ–љ–Є–µ —Н—В–∞–љ–Њ–ї–∞ –≤–µ–і–µ—В –Ї –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—О –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е —Б–≤–Њ–±–Њ–і–љ—Л—Е —А–∞–і–Є–Ї–∞–ї–Њ–≤ –≤ –њ–µ—З–µ–љ–Є. –Ъ –љ–Є–Љ –Њ—В–љ–Њ—Б—П—В—Б—П –≥–Є–і—А–Њ–Ї—Б–Є—Н—В–Є–ї–Њ–≤—Л–є —А–∞–і–Є–Ї–∞–ї, —Б—Г–њ–µ—А–Њ–Ї—Б–Є–і –∞–љ–Є–Њ–љ (–Ю2–) –Є –≥–Є–і—А–Њ–Ї—Б–Є–ї—М–љ—Л–є —А–∞–і–Є–Ї–∞–ї (–Ю–Э–). –≠—В–Є —В—А–Є —А–∞–і–Є–Ї–∞–ї–∞ –≤—Л–Ј—Л–≤–∞—О—В —А–∞–Ј–ї–Є—З–љ—Л–µ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ—Л—Е –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤. –Ъ–ї–∞—Б—Б–Є—З–µ—Б–Ї–Є –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ —А–∞–і–Є–Ї–∞–ї–Њ–≤ —Б–≤—П–Ј—Л–≤–∞—О—В —Б –Њ–Ї–Є—Б–ї–µ–љ–Є–µ–Љ —Н—В–∞–љ–Њ–ї–∞ —Б–Є—Б—В–µ–Љ–Њ–є —Ж–Є—В–Њ—Е—А–Њ–Љ–∞ CYP2E1. –Ю–і–љ–∞–Ї–Њ —Н—В–Њ –љ–µ —Б–Њ–≤—Б–µ–Љ —В–∞–Ї. –С—Л–ї–Њ –њ—А–Њ–≤–µ–і–µ–љ–Њ –Њ–і–љ–Њ –≤–µ—Б—М–Љ–∞ –Є–љ—В–µ—А–µ—Б–љ–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ, –≥–і–µ –Љ—Л—И–∞–Љ, —Г –Ї–Њ—В–Њ—А—Л—Е –Њ—В—Б—Г—В—Б—В–≤–Њ–≤–∞–ї CYP2E1, –≤–≤–Њ–і–Є–ї–Є —Н—В–∞–љ–Њ–ї. –Ю–і–љ–∞–Ї–Њ –≤ –њ–µ—З–µ–љ–Є —Н—В–Є—Е –Љ—Л—И–µ–є –њ—А–Њ–і–Њ–ї–ґ–∞–ї–Є –Њ–±—А–∞–Ј–Њ–≤—Л–≤–∞—В—М—Б—П —Б–≤–Њ–±–Њ–і–љ—Л–µ —А–∞–і–Є–Ї–∞–ї—Л, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞ –њ–Њ–ї–љ–Њ–µ –Њ—В—Б—Г—В—Б—В–≤–Є–µ CYP2E1 [5]. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –і–∞–љ–љ–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Њ –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В—М, —З—В–Њ –≤ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–Є —Б–≤–Њ–±–Њ–і–љ—Л—Е —А–∞–і–Є–Ї–∞–ї–Њ–≤ –≤ –њ–µ—З–µ–љ–Є –њ–Њ–і –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ–Љ —Н—В–∞–љ–Њ–ї–∞ –≤–Њ–≤–ї–µ—З–µ–љ –љ–µ –Њ–і–Є–љ –Љ–µ—Е–∞–љ–Є–Ј–Љ –Є –±–Њ–ї–µ–µ —З–µ–Љ –Њ–і–Є–љ —В–Є–њ –Ї–ї–µ—В–Њ–Ї. –Ъ —В–∞–Ї–Є–Љ –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞–Љ –Љ–Њ–ґ–љ–Њ –Њ—В–љ–µ—Б—В–Є –Љ–Њ–±–Є–ї–Є–Ј–∞—Ж–Є—О –ґ–µ–ї–µ–Ј–∞ –Є–Ј —Д–µ—А—А–Є—В–Є–љ–∞ –њ–Њ–і –і–µ–є—Б—В–≤–Є–µ–Љ –Є–Ј–±—Л—В–Ї–∞ –Э–Р–Ф–Э –Є —Г—З–∞—Б—В–Є–µ —А–∞–Ј–ї–Є—З–љ—Л—Е —В–Є–њ–Њ–≤ –Ї–ї–µ—В–Њ–Ї (–ї–µ–є–Ї–Њ—Ж–Є—В–Њ–≤, –љ–µ–є—В—А–Њ—Д–Є–ї–Њ–≤, –∞ —В–∞–Ї–ґ–µ –Ъ—Г–њ—Д—Д–µ—А–Њ–≤—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї) [2,3].
–°–≤–Њ–±–Њ–і–љ—Л–µ —А–∞–і–Є–Ї–∞–ї—Л –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г—О—В —Б –љ–µ–љ–∞—Б—Л—Й–µ–љ–љ—Л–Љ–Є –ї–Є–њ–Є–і–∞–Љ–Є –Є –Ј–∞–њ—Г—Б–Ї–∞—О—В –њ–µ—А–µ–Ї–Є—Б–љ–Њ–µ –Њ–Ї–Є—Б–ї–µ–љ–Є–µ –ї–Є–њ–Є–і–Њ–≤. –•—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ —Н—В–∞–љ–Њ–ї–∞ –≤–µ–і–µ—В –Ї –њ–µ—А–µ–Ї–Є—Б–љ–Њ–Љ—Г –Њ–Ї–Є—Б–ї–µ–љ–Є—О –ї–Є–њ–Є–і–Њ–≤ –≤ –њ–µ—З–µ–љ–Є, –Ї–Њ—В–Њ—А–Њ–µ, –≤ —Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –≤—Л–Ј—Л–≤–∞–µ—В –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ —В–Ї–∞–љ–µ–є –Є —А–∞–Ј–≤–Є—В–Є–µ —Д–Є–±—А–Њ–Ј–∞. –°–≤–Њ–±–Њ–і–љ—Л–µ —А–∞–і–Є–Ї–∞–ї—Л —В–∞–Ї–ґ–µ –њ–Њ–≤—А–µ–ґ–і–∞—О—В –Є –Ї–ї–µ—В–Њ—З–љ—Г—О –Ф–Э–Ъ. –Ь–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–∞—П –Ф–Э–Ъ –±–Њ–ї–µ–µ —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–∞ –Ї –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ–Њ–Љ—Г –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О, —З–µ–Љ —П–і–µ—А–љ–∞—П –Ф–Э–Ъ, —В–∞–Ї –Ї–∞–Ї –≤ –Љ–µ–љ—М—И–µ–є —Б—В–µ–њ–µ–љ–Є –Ј–∞—Й–Є—Й–µ–љ–∞ –≥–Є—Б—В–Њ–љ–Њ–≤—Л–Љ–Є –Є –љ–µ–≥–Є—Б—В–Њ–љ–Њ–≤—Л–Љ–Є –±–µ–ї–Ї–∞–Љ–Є, –∞ —В–∞–Ї–ґ–µ –Є–Ј––Ј–∞ –Љ–µ–љ—М—И–µ–є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є –Ї —А–µ–њ–∞—А–∞—Ж–Є–Є. –Ю–Ї—Б–Є–і–∞–љ—В—Л –Љ–Њ–≥—Г—В –≤—Л–Ј—Л–≤–∞—В—М –Љ—Г—В–∞—Ж–Є–Є –≤ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–Њ–є –Ф–Э–Ъ, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П–Љ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є. –≠—Д—Д–µ–Ї—В—Л —Б–≤–Њ–±–Њ–і–љ—Л—Е —А–∞–і–Є–Ї–∞–ї–Њ–≤ –љ–∞ –њ–µ—З–µ–љ—М —Г—Б–Є–ї–Є–≤–∞—О—В—Б—П –Є–Ј––Ј–∞ —В–Њ–≥–Њ, —З—В–Њ —Н—В–∞–љ–Њ–ї –њ—А–Є–≤–Њ–і–Є—В —В–∞–Ї–ґ–µ –Є –Ї –љ–∞—А—Г—И–µ–љ–Є—О –∞–љ—В–Є–Њ–Ї—Б–Є–і–∞–љ—В–љ–Њ–є –Ј–∞—Й–Є—В—Л. –•—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ —Н—В–∞–љ–Њ–ї–∞ –њ—А–Є–≤–Њ–і–Є—В –Ї —Г–Љ–µ–љ—М—И–µ–љ–Є—О –≤–Є—В–∞–Љ–Є–љ–Њ–≤ –Р –Є –Х, –∞ —В–∞–Ї–ґ–µ –≥–ї—Г—В–∞—В–Є–Њ–љ–∞. –Я—А–Є –і–µ—Д–Є—Ж–Є—В–µ –≤–Є—В–∞–Љ–Є–љ–∞ –Х —Г—Б–Є–ї–Є–≤–∞–µ—В—Б—П –њ–µ—А–µ–Ї–Є—Б–љ–Њ–µ –Њ–Ї–Є—Б–ї–µ–љ–Є–µ –ї–Є–њ–Є–і–Њ–≤. –Э–µ—Е–≤–∞—В–Ї–∞ –≤–Є—В–∞–Љ–Є–љ–∞ –Р —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О –ї–Є–Ј–Њ—Б–Њ–Љ, –∞ —Б–љ–Є–ґ–µ–љ–Є–µ —Б–Њ–і–µ—А–ґ–∞–љ–Є—П –≥–ї—Г—В–∞—В–Є–Њ–љ–∞ –≤–µ–і–µ—В –Ї –љ–∞—А—Г—И–µ–љ–Є—О —Д—Г–љ–Ї—Ж–Є–Є –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –Є –і–µ–ї–∞–µ—В –Ї–ї–µ—В–Ї—Г –±–Њ–ї–µ–µ —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ–є –Ї –∞–њ–Њ–њ—В–Њ–Ј—Г [3,4].
–У–Є–њ–Њ–Ї—Б–Є—П. –•—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ –∞–ї–Ї–Њ–≥–Њ–ї—П –≤—Л–Ј—Л–≤–∞–µ—В –≥–Є–њ–µ—А–Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Њ–µ —Б–Њ—Б—В–Њ—П–љ–Є–µ –њ–µ—З–µ–љ–Є —Б –њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ –њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ–Љ –Ї–Є—Б–ї–Њ—А–Њ–і–∞ –Ї–ї–µ—В–Ї–∞–Љ–Є –њ–µ—З–µ–љ–Є. –≠—В–Њ —Б–Њ—Б—В–Њ—П–љ–Є–µ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В –њ–Њ—А—В–Њ–—Ж–µ–љ—В—А–∞–ї—М–љ—Л–є –Ї–Є—Б–ї–Њ—А–Њ–і–љ—Л–є –≥—А–∞–і–Є–µ–љ—В, –Њ—Б—В–∞–≤–ї—П—П –њ–µ—А–Є—Ж–µ–љ—В—А–∞–ї—М–љ—Л–µ –≥–µ–њ–∞—В–Њ—Ж–Є—В—Л –≤ —Б–Њ—Б—В–Њ—П–љ–Є–Є –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ–є –≥–Є–њ–Њ–Ї—Б–Є–Є. –Т—Б–µ —Н—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї –Є—Б—В–Њ—Й–µ–љ–Є—О –Р–Ґ–§ –Є –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О –њ–µ—З–µ–љ–Є [3].
–Ф–µ–є—Б—В–≤–Є–µ –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і–∞. –•–Њ—В—П –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і –≤ –љ–Њ—А–Љ–µ –±—Л—Б—В—А–Њ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Є—А—Г–µ—В—Б—П –≤ –њ–µ—З–µ–љ–Є –і–Њ –∞—Ж–µ—В–∞—В–∞, —Г –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤ –Њ–љ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –Ј–∞–Љ–µ–і–ї–µ–љ, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—О –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і–∞. –Я—А–Є –і–Њ—Б—В–Є–ґ–µ–љ–Є–Є –Њ–њ—А–µ–і–µ–ї–µ–љ–љ–Њ–є, –і–Њ—Б—В–∞—В–Њ—З–љ–Њ –≤—Л—Б–Њ–Ї–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Њ–љ —Б—В–∞–љ–Њ–≤–Є—В—Б—П —Б—Г–±—Б—В—А–∞—В–Њ–Љ –Ї—Б–∞–љ—В–Є–љ –Њ–Ї—Б–Є–і–∞–Ј—Л –Є –∞–ї—М–і–µ–≥–Є–і –Њ–Ї—Б–Є–і–∞–Ј—Л, –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —З–µ–≥–Њ –Њ–±—А–∞–Ј—Г—О—В—Б—П —Б–≤–Њ–±–Њ–і–љ—Л–µ —А–∞–і–Є–Ї–∞–ї—Л (—А–Є—Б. 1). –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і –љ–∞—А—Г—И–∞–µ—В –±–µ—В–∞––Њ–Ї–Є—Б–ї–µ–љ–Є–µ –ґ–Є—А–љ—Л—Е –Ї–Є—Б–ї–Њ—В –≤ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є—П—Е –Є –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г–µ—В —Б–Њ —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–Љ–Є –∞–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В–љ—Л–Љ–Є –Њ—Б—В–∞—В–Ї–∞–Љ–Є –љ–∞ –Ї–ї–µ—В–Њ—З–љ—Л—Е –±–µ–ї–Ї–∞—Е —Б –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ–Љ –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і––±–µ–ї–Ї–Њ–≤—Л—Е –њ–Њ–±–Њ—З–љ—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤. –≠—В–Є –њ—А–Њ–і—Г–Ї—В—Л —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В —А–∞–Ј–≤–Є—В–Є—О –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є –Ј–∞ —Б—З–µ—В –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ –≤—Л–Ј—Л–≤–∞—О—В –Є–Љ–Љ—Г–љ–љ—Л–є –Њ—В–≤–µ—В –Є —Г—Б–Є–ї–Є–≤–∞—О—В —Б–Є–љ—В–µ–Ј –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ –≤ –њ–µ—З–µ–љ–Є. –Р—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і –Љ–Њ–ґ–µ—В –≤—Б—В—Г–њ–∞—В—М –≤ —А–µ–∞–Ї—Ж–Є–Є —Б —В–∞–Ї–Є–Љ–Є –±–µ–ї–Ї–∞–Љ–Є, –Ї–∞–Ї —В—Г–±—Г–ї–Є–љ, –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ –≤—Л–Ј—Л–≤–∞—П –љ–∞—А—Г—И–µ–љ–Є–µ –Њ–±—К–µ–і–Є–љ–µ–љ–Є—П –Љ–Є–Ї—А–Њ—В—А—Г–±–Њ—З–µ–Ї, —З—В–Њ, –≤ —Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –њ–Њ–≤—А–µ–ґ–і–∞–µ—В —В—А–∞–љ—Б–њ–Њ—А—В–љ—Л–µ –њ—А–Њ—Ж–µ—Б—Б—Л –≤ –≥–µ–њ–∞—В–Њ—Ж–Є—В–µ –Є –љ–∞—А—Г—И–∞–µ—В —Б–Є–љ—В–µ–Ј –±–µ–ї–Ї–∞. –Ш—В–Њ–≥ —Н—В–Є—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є – –±–∞–ї–ї–Њ–љ–љ–∞—П –і–Є–ї–∞—В–∞—Ж–Є—П –≥–µ–њ–∞—В–Њ—Ж–Є—В–Њ–≤ – —Е–∞—А–∞–Ї—В–µ—А–љ—Л–є –њ—А–Є–Ј–љ–∞–Ї –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є [2].
–Ш–Љ–Љ—Г–љ–љ—Л–µ –Є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –њ–µ—З–µ–љ–Є. –Ъ—Г–њ—Д—Д–µ—А–Њ–≤—Б–Ї–Є–µ –Ї–ї–µ—В–Ї–Є, –Ї–Њ—В–Њ—А—Л–µ, –њ–Њ —Б—Г—В–Є, —П–≤–ї—П—О—В—Б—П –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–Љ–Є –њ–µ—З–µ–љ–Є, –≤—Л—А–∞–±–∞—В—Л–≤–∞—О—В –Њ–Ї—Б–Є–і–∞–љ—В—Л –Є —Ж–Є—В–Њ–Ї–Є–љ—Л, –≤ –љ–Њ—А–Љ–µ –≤—Л–њ–Њ–ї–љ—П—О—Й–Є–µ –Ј–∞—Й–Є—В–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є. –•—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ –∞–ї–Ї–Њ–≥–Њ–ї—П –≤—Л–Ј—Л–≤–∞–µ—В –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї—Г—О –∞–Ї—В–Є–≤–∞—Ж–Є—О –Ъ—Г–њ—Д—Д–µ—А–Њ–≤—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї. –Я—А–Є —Н—В–Њ–Љ —Ж–Є—В–Њ–Ї–Є–љ—Л –Є –Њ–Ї—Б–Є–і–∞–љ—В—Л, –≤—Л—Б–≤–Њ–±–Њ–ґ–і–∞—О—Й–Є–µ—Б—П –Є–Ј —Н—В–Є—Е –Ї–ї–µ—В–Њ–Ї, –≤—Л–Ј—Л–≤–∞—О—В –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –њ–µ—З–µ–љ–Є. –¶–µ–љ—В—А–∞–ї—М–љ—Г—О —А–Њ–ї—М –Ъ—Г–њ—Д—Д–µ—А–Њ–≤—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї –≤ –њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є –і–Њ–Ї–∞–Ј–∞–ї–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –њ—А–Њ–≤–µ–і–µ–љ–љ—Л–µ –љ–∞ –ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л—Е –ґ–Є–≤–Њ—В–љ—Л—Е. –Я—А–Є –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ј–∞—Ж–Є–Є –Ї—А—Л—Б, –ї–Є—И–µ–љ–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ —Б–њ–Њ—Б–Њ–±–љ—Л—Е –Ъ—Г–њ—Д—Д–µ—А–Њ–≤—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї, –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –њ–µ—З–µ–љ–Є —Г –љ–Є—Е –љ–µ –≤–Њ–Ј–љ–Є–Ї–∞–ї–Њ. –°—А–µ–і–Є –Љ–љ–Њ–ґ–µ—Б—В–≤–∞ –≤–µ—Й–µ—Б—В–≤, –≤—Л—А–∞–±–∞—В—Л–≤–∞–µ–Љ—Л—Е –∞–Ї—В–Є–≤–Є—А–Њ–≤–∞–љ–љ—Л–Љ–Є –Ъ—Г–њ—Д—Д–µ—А–Њ–≤—Б–Ї–Є–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є – —Д–∞–Ї—В–Њ—А –љ–µ–Ї—А–Њ–Ј–∞ –Њ–њ—Г—Е–Њ–ї–Є – α (–§–Э–Ю–α), —В—А–∞–љ—Б—Д–Њ—А–Љ–Є—А—Г—О—Й–Є–є —Д–∞–Ї—В–Њ—А —А–Њ—Б—В–∞ – β (–Ґ–§–†–β) –Є —Б—Г–њ–µ—А–Њ–Ї—Б–Є–і, –њ–Њ –≤—Б–µ–є –≤–Є–і–Є–Љ–Њ—Б—В–Є, –Є–≥—А–∞—О—В –≥–ї–∞–≤–љ—Г—О —А–Њ–ї—М –≤ –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–Є –њ–µ—З–µ–љ–Є. –Я—А–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–Љ —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–Є —Н—В–∞–љ–Њ–ї–∞ –§–Э–Ю —Г—Б–Є–ї–Є–≤–∞–µ—В –∞–њ–Њ–њ—В–Њ–Ј –≥–µ–њ–∞—В–Њ—Ж–Є—В–Њ–≤. –§–Э–Ю —В–∞–Ї–ґ–µ –≤—Л–Ј—Л–≤–∞–µ—В —Н–Ї—Б–њ—А–µ—Б—Б–Є—О –≤ –њ–µ—З–µ–љ–Є –Љ–љ–Њ–≥–Є—Е —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤ –Є –Љ–Њ–ї–µ–Ї—Г–ї –Ї–ї–µ—В–Њ—З–љ–Њ–є –∞–і–≥–µ–Ј–Є–Є, –Ї–Њ—В–Њ—А—Л–µ –њ—А–Є–≤–ї–µ–Ї–∞—О—В –ї–µ–є–Ї–Њ—Ж–Є—В—Л –≤ –њ–µ—З–µ–љ—М. –°—А–µ–і–Є —Н—В–Є—Е —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤ –Є –Љ–Њ–ї–µ–Ї—Г–ї – –Ш–Ы–1, –Ш–Ы–6, –Ш–Ы–8, –Љ–Њ–љ–Њ—Ж–Є—В–∞—А–љ—Л–є —Е–µ–Љ–Њ–∞—В—В—А–∞–Ї—В–∞–љ—В–љ—Л–є –њ—А–Њ—В–µ–Є–љ–1, –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–ї—М–љ—Л–є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–є –±–µ–ї–Њ–Ї–1α, 1β –Є –Њ–љ–Ї–Њ–≥–µ–љ —А–Њ—Б—В–∞ –∞–ї—М—Д–∞. –≠—В–Є –њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ —Ж–Є—В–Њ–Ї–Є–љ—Л –њ—А–Є–≤–Њ–і—П—В –Ї —Б–Є–љ–і—А–Њ–Љ—Г –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–≥–Њ –≥–µ–њ–∞—В–Є—В–∞. –£—А–Њ–≤–љ–Є –§–Э–Ю –Є –Ш–Ы–8 –≤ –Ї—А–Њ–≤–Є –Ї–Њ—А—А–µ–ї–Є—А—Г—О—В —Б —В—П–ґ–µ—Б—В—М—О –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Т—Л—А–∞–±–∞—В—Л–≤–∞–µ–Љ—Л–є –Ї–ї–µ—В–Ї–∞–Љ–Є –Ъ—Г–њ—Д—Д–µ—А–∞ —Б—Г–њ–µ—А–Њ–Ї—Б–Є–і –Њ–±–ї–∞–і–∞–µ—В —Б–≤–Њ–є—Б—В–≤–∞–Љ–Є —Б—А–∞–≤–љ–Є–Љ—Л–Љ–Є —Б –§–Э–Ю. –°—Г–њ–µ—А–Њ–Ї—Б–Є–і –∞–Ї—В–Є–≤–Є—А—Г–µ—В —П–і–µ—А–љ—Л–є —В—А–∞–љ—Б–Ї—А–Є–њ—Ж–Є–Њ–љ–љ—Л–є —Д–∞–Ї—В–Њ—А NF–κβ, –Ї–Њ—В–Њ—А—Л–є –љ–∞–њ—А—П–Љ—Г—О —А–µ–≥—Г–ї–Є—А—Г–µ—В —В—А–∞–љ—Б–Ї—А–Є–њ—Ж–Є—О –≥–µ–љ–Њ–≤, –Ї–Њ–і–Є—А—Г—О—Й–Є—Е —Ж–Є—В–Њ–Ї–Є–љ—Л –Є –Љ–Њ–ї–µ–Ї—Г–ї—Л –Ї–ї–µ—В–Њ—З–љ–Њ–є –∞–і–≥–µ–Ј–Є–Є [4].
–Х—Б–ї–Є –≥–µ–њ–∞—В–Њ—Ж–µ–ї–ї—О–ї—П—А–љ—Л–µ –±–µ–ї–Ї–Є –Њ–±—А–∞–Ј—Г—О—В –њ–Њ–±–Њ—З–љ—Л–µ –њ—А–Њ–і—Г–Ї—В—Л —Б –∞–ї—М–і–µ–≥–Є–і–Њ–Љ –Є–ї–Є –≥–Є–і—А–Њ–Ї—Б–Є—Н—В–Є–ї–Њ–≤—Л–Љ —А–∞–і–Є–Ї–∞–ї–Њ–Љ, —В–Њ –њ–Њ–і–Њ–±–љ—Л–µ —Б–Њ–µ–і–Є–љ–µ–љ–Є—П –Љ–Њ–≥—Г—В –≤—Л–Ј–≤–∞—В—М –Є–Љ–Љ—Г–љ–љ—Л–є –Њ—В–≤–µ—В. –Р–љ—В–Є—В–µ–ї–∞, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л–µ –њ—А–Њ—В–Є–≤ —В–∞–Ї–Є—Е –Є–Ј–Љ–µ–љ–µ–љ–љ—Л—Е –±–µ–ї–Ї–Њ–≤, –Њ–њ—А–µ–і–µ–ї—П—О—В—Б—П –≤ –Ї—А–Њ–≤–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ—М—О –њ–µ—З–µ–љ–Є. –Ю–і–љ–∞–Ї–Њ —А–Њ–ї—М —Н—В–Є—Е –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї –≤ –њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–Њ–Ї–∞ –љ–µ —П—Б–љ–∞ [6].
–Ь–µ—Е–∞–љ–Є–Ј–Љ—Л —Д–Є–±—А–Њ–Ј–∞. –§–Є–±—А–Њ–Ј –њ–µ—З–µ–љ–Є – —В—П–ґ–µ–ї–Њ–µ –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ–Њ –љ–µ–Њ–±—А–∞—В–Є–Љ–Њ–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є–µ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є—П —Н—В–∞–љ–Њ–ї–∞. –§–Є–±—А–Њ–Ј —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Г 10–15% –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤, –љ–Њ –њ–Њ—З—В–Є —Г 50% —Б –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є —П–≤–љ–Њ–є –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ—М—О –њ–µ—З–µ–љ–Є. –¶–µ–љ—В—А–∞–ї—М–љ—Л–Љ –њ–∞—В–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–Љ –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є —П–≤–ї—П–µ—В—Б—П –∞–Ї—В–Є–≤–∞—Ж–Є—П –Ј–≤–µ–Ј–і—З–∞—В—Л—Е –Ї–ї–µ—В–Њ–Ї. –Ч–≤–µ–Ј–і—З–∞—В—Л–µ –Ї–ї–µ—В–Ї–Є —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ—Л –≤ –њ—А–Њ—Б—В—А–∞–љ—Б—В–≤–∞—Е –Ф–Є—Б—Б–µ –Љ–µ–ґ–і—Г –≥–µ–њ–∞—В–Њ—Ж–Є—В–Њ–Љ –Є —Н–љ–і–Њ—В–µ–ї–Є–µ–Љ —Б–Є–љ—Г—Б–Њ–Є–і–∞. –Т –љ–Њ—А–Љ–µ –Ј–≤–µ–Ј–і—З–∞—В—Л–µ –Ї–ї–µ—В–Ї–Є –љ–∞—Е–Њ–і—П—В—Б—П –≤ —Б–Њ—Б—В–Њ—П–љ–Є–Є –њ–Њ–Ї–Њ—П –Є —П–≤–ї—П—О—В—Б—П –Љ–µ—Б—В–Њ–Љ —Е—А–∞–љ–µ–љ–Є—П –≤–Є—В–∞–Љ–Є–љ–∞ –Р –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ. –Я—А–Є –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–Є –њ–µ—З–µ–љ–Є, –≤—Л–Ј–≤–∞–љ–љ–Њ–Љ –Ї–∞–Ї —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ–Љ –∞–ї–Ї–Њ–≥–Њ–ї—П, —В–∞–Ї –Є –і—А—Г–≥–Є–Љ–Є —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Є–Љ–Є –Є–ї–Є –≤–Є—А—Г—Б–љ—Л–Љ–Є –∞–≥–µ–љ—В–∞–Љ–Є, –Ј–≤–µ–Ј–і—З–∞—В—Л–µ –Ї–ї–µ—В–Ї–Є –љ–∞—З–Є–љ–∞—О—В –њ—А–Њ–ї–Є—Д–µ—А–Є—А–Њ–≤–∞—В—М –Є –њ—А–µ–≤—А–∞—Й–∞—О—В—Б—П –≤ –Љ–Є–Њ—Д–Є–±—А–Њ–±–ї–∞—Б—В–њ–Њ–і–Њ–±–љ—Л–µ –Ї–ї–µ—В–Ї–Є. –Р–Ї—В–Є–≤–Є—А–Њ–≤–∞–љ–љ—Л–µ –Ј–≤–µ–Ј–і—З–∞—В—Л–µ –Ї–ї–µ—В–Ї–Є –љ–∞—З–Є–љ–∞—О—В –≤—Л—А–∞–±–∞—В—Л–≤–∞—В—М –Ї–Њ–ї–ї–∞–≥–µ–љ. –Т —А–µ–Ј—Г–ї—М—В–∞—В–µ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –њ–µ—А–Є—Б–Є–љ—Г—Б–Њ–Є–і–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–є –і–ї—П –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є [2,3].
–Ґ–Њ—З–љ—Л–µ —Б—В–Є–Љ—Г–ї—Л –∞–Ї—В–Є–≤–∞—Ж–Є–Є –Ј–≤–µ–Ј–і—З–∞—В—Л—Е –Ї–ї–µ—В–Њ–Ї –љ–µ –Є–Ј–≤–µ—Б—В–љ—Л. –Я—А–Є –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є —В–∞–Ї–Є–Љ–Є «—Б—В–Є–Љ—Г–ї–∞–Љ–Є» –Љ–Њ–≥—Г—В —П–≤–ї—П—В—М—Б—П –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і –Є –∞–ї—М–і–µ–≥–Є–і––±–µ–ї–Ї–Њ–≤—Л–µ –њ—А–Њ–Є–Ј–≤–Њ–і–љ—Л–µ, –∞ —В–∞–Ї–ґ–µ –Њ–Ї—Б–Є–і–∞–љ—В—Л –Є –њ—А–Њ–і—Г–Ї—В—Л –њ–µ—А–µ–Ї–Є—Б–љ–Њ–≥–Њ –Њ–Ї–Є—Б–ї–µ–љ–Є—П –ї–Є–њ–Є–і–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ –∞–Ї—В–Є–≤–Є—А—Г—О—В –Ј–≤–µ–Ј–і—З–∞—В—Л–µ –Ї–ї–µ—В–Ї–Є –Є —Б—В–Є–Љ—Г–ї–Є—А—Г—О—В —Б–Є–љ—В–µ–Ј –Ї–Њ–ї–ї–∞–≥–µ–љ–∞. –Т–∞–ґ–љ—Г—О —А–Њ–ї—М –≤ —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–Є —Д–Є–±—А–Њ–Ј–∞ –њ–µ—З–µ–љ–Є –Є–≥—А–∞–µ—В –Ґ–§–†––±–µ—В–∞, –Ї–Њ—В–Њ—А—Л–є, –Ї–∞–Ї –±—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —Б–њ–Њ—Б–Њ–±–µ–љ –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞—В—М —Б–Є–љ—В–µ–Ј –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ –Ј–≤–µ–Ј–і—З–∞—В—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є –≤ –Ї—Г–ї—М—В—Г—А–µ –Ї–ї–µ—В–Њ–Ї [7].
–§–∞–Ї—В–Њ—А—Л —А–Є—Б–Ї–∞
–∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є
–Э–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ—Б—В—М. –Ґ–∞–Ї –Ї–∞–Ї –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –њ–µ—З–µ–љ–Є —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –ї–Є—И—М —Г –љ–µ–±–Њ–ї—М—И–Њ–є —З–∞—Б—В–Є –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤, —В–Њ –Њ—З–µ–≤–Є–і–љ–Њ, —З—В–Њ –љ–µ —В–Њ–ї—М–Ї–Њ —Б–∞–Љ —Н—В–∞–љ–Њ–ї, –љ–Њ –Є –і—А—Г–≥–Є–µ —Д–∞–Ї—В–Њ—А—Л –Є–≥—А–∞—О—В —А–Њ–ї—М –≤ –њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –±–Њ–ї–µ–Ј–љ–Є, –≤ —З–∞—Б—В–љ–Њ—Б—В–Є, –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ—Б—В—М. –Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —Б—А–µ–і–Є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤ –њ—А–µ–і—А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ–Њ—Б—В–Є –Ї –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–Љ—Г –њ–Њ—А–∞–ґ–µ–љ–Є—О –њ–µ—З–µ–љ–Є –Њ—Б–љ–Њ–≤–љ—Г—О —А–Њ–ї—М –Њ—В–≤–Њ–і—П—В –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ—Г –±–µ–ї–Ї–Њ–≤, —Г—З–∞—Б—В–≤—Г—О—Й–Є—Е –≤ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–µ –∞–ї–Ї–Њ–≥–Њ–ї—П –Є —А–µ–≥—Г–ї–Є—А—Г—О—Й–Є—Е –Є–Љ–Љ—Г–љ–љ—Л–є –Њ—В–≤–µ—В –Њ—А–≥–∞–љ–Є–Ј–Љ–∞. –Я–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ –≥–µ–љ–Њ–≤, –Ї–Њ–і–Є—А—Г—О—Й–Є—Е –∞–ї–Ї–Њ–≥–Њ–ї—М–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј—Г 2 —В–Є–њ–∞, –∞–ї—М–і–µ–≥–Є–і–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј—Г 2 —В–Є–њ–∞ –Є —Ж–Є—В–Њ—Е—А–Њ–Љ 2–Х1, –Љ–Њ–ґ–µ—В –Њ–њ—А–µ–і–µ–ї—П—В—М —А–Є—Б–Ї —А–∞–Ј–≤–Є—В–Є—П –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є. –Ф—А—Г–≥–Є–Љ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–Љ –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–Љ, –Њ–±—К—П—Б–љ—П—О—Й–Є–Љ –≥–µ–љ–µ—В–Є—З–µ—Б–Ї—Г—О –њ—А–µ–і—А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ–Њ—Б—В—М –Ї –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–Љ—Г –њ–Њ—А–∞–ґ–µ–љ–Є—О –њ–µ—З–µ–љ–Є, —П–≤–ї—П—О—В—Б—П –і–∞–љ–љ—Л–µ –Њ —А–Њ–ї–Є –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–∞ –±–µ–ї–Ї–Њ–≤, —А–µ–≥—Г–ї–Є—А—Г—О—Й–Є—Е –Є–Љ–Љ—Г–љ–љ—Л–є –Њ—В–≤–µ—В –Њ—А–≥–∞–љ–Є–Ј–Љ–∞, –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ —Н–љ–і–Њ—В–Њ–Ї—Б–Є–љ–Њ–≤ –љ–∞ –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–Є –Ї–ї–µ—В–Њ–Ї –Ъ—Г–њ—Д—Д–µ—А–∞, —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤ –Є –Є—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤. –Я–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ –≥–µ–љ–Њ–≤, –Ї–Њ–і–Є—А—Г—О—Й–Є—Е –±–µ–ї–Ї–Є –°D14, –Ш–Ы–10, –§–Э–Ю –Є CTLA–4, –Њ–њ—А–µ–і–µ–ї—П–µ—В –њ–Њ–≤—Л—И–µ–љ–љ—Г—О —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В—М –Њ—А–≥–∞–љ–Є–Ј–Љ–∞ –Ї –∞–ї–Ї–Њ–≥–Њ–ї—О [8].
–Я–Њ–ї. –Ц–µ–љ—Й–Є–љ—Л –≤ –±–Њ–ї—М—И–µ–є —Б—В–µ–њ–µ–љ–Є –њ–Њ–і–≤–µ—А–ґ–µ–љ—Л –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–Љ—Г –њ–Њ—А–∞–ґ–µ–љ–Є—О –њ–µ—З–µ–љ–Є, —З–µ–Љ –Љ—Г–ґ—З–Є–љ—Л. –Ґ–∞–Ї–ґ–µ —Г –ґ–µ–љ—Й–Є–љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ —Б–Ї–ї–Њ–љ–љ–Њ –Ї –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—О –і–∞–ґ–µ –њ—А–Є –∞–±—Б—В–Є–љ–µ–љ—Ж–Є–Є. –Я–Њ–ї–Њ–≤—Л–µ —А–∞–Ј–ї–Є—З–Є—П –≤ –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –љ–µ–Њ–±—К—П—Б–љ–Є–Љ—Л. –Я—А–µ–і–њ–Њ–ї–∞–≥–∞—О—В, —З—В–Њ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ —Н—В–Њ —Б–≤—П–Ј–∞–љ–Њ —Б –±–Њ–ї–µ–µ –љ–Є–Ј–Ї–Є–Љ —Г—А–Њ–≤–љ–µ–Љ –ґ–µ–ї—Г–і–Њ—З–љ–Њ–є –∞–ї–Ї–Њ–≥–Њ–ї—М–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј—Л —Г –ґ–µ–љ—Й–Є–љ. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, —Б—Г—Й–µ—Б—В–≤—Г–µ—В —В–µ–Њ—А–Є—П —Г—Б–Ї–Њ—А–µ–љ–љ–Њ–≥–Њ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –њ–µ—З–µ–љ–Є —Г –ґ–µ–љ—Й–Є–љ, –Њ—Б–љ–Њ–≤–∞–љ–љ–∞—П –љ–∞ –њ–Њ–ї–Њ–≤—Л—Е —А–∞–Ј–ї–Є—З–Є—П—Е –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ –ґ–Є—А–љ—Л—Е –Ї–Є—Б–ї–Њ—В. –Х—Б–ї–Є –ґ–Є—А–љ—Л–µ –Ї–Є—Б–ї–Њ—В—Л –љ–∞–Ї–∞–њ–ї–Є–≤–∞—О—В—Б—П –≤ –Ї–ї–µ—В–Ї–∞—Е –њ–µ—З–µ–љ–Є –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ –љ–∞—А—Г—И–µ–љ–љ–Њ–≥–Њ –±–µ—В–∞––Њ–Ї–Є—Б–ї–µ–љ–Є—П, –љ–µ –њ—А–µ–≤—А–∞—Й–∞—П—Б—М –≤ —В—А–Є–≥–ї–Є—Ж–µ—А–Є–і—Л, –Њ–љ–Є –Љ–Њ–≥—Г—В –≤—Л–Ј—Л–≤–∞—В—М –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –њ–µ—З–µ–љ–Є. –Я—А–Њ–Є—Б—Е–Њ–і–Є—В —Н—В–Њ –Є–Ј––Ј–∞ –∞–ї—М—В–µ—А–љ–∞—В–Є–≤–љ—Л—Е –њ—Г—В–µ–є –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞, –≤ —З–∞—Б—В–љ–Њ—Б—В–Є, —В–∞–Ї–Њ–≥–Њ –Ї–∞–Ї —Ж–Є—В–Њ—Е—А–Њ–Љ –†450–Р1––Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ–≥–Њ –Њ–Љ–µ–≥–∞ –≥–Є–і—А–Њ–Ї—Б–Є–ї–Є—А–Њ–≤–∞–љ–Є—П. –≠—В–Њ—В –Ї–Њ–Љ–њ–µ–љ—Б–∞—В–Њ—А–љ—Л–є –њ—Г—В—М –∞–Ї—В–Є–≤–љ–Њ —А–∞–±–Њ—В–∞–µ—В —Г –Љ—Г–ґ—Б–Ї–Є—Е –Њ—Б–Њ–±–µ–є –Ї—А—Л—Б, –∞ —Г –ґ–µ–љ—Б–Ї–Є—Е –µ–≥–Њ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Б–љ–Є–ґ–µ–љ–∞. –°–≤—П–Ј—Л–≤–∞—О—Й–∞—П —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –ґ–Є—А–љ—Л—Е –Ї–Є—Б–ї–Њ—В —В–∞–Ї–ґ–µ —Б–љ–Є–ґ–µ–љ–∞ —Г –ґ–µ–љ—Б–Ї–Є—Е –Њ—Б–Њ–±–µ–є –Ї—А—Л—Б –њ–Њ—Б–ї–µ –і–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –∞–ї–Ї–Њ–≥–Њ–ї—П. –°–љ–Є–ґ–µ–љ–Є–µ —Б–≤—П–Ј—Л–≤–∞—О—Й–µ–є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є –Љ–Њ–ґ–µ—В —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞—В—М —В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–Љ—Г –і–µ–є—Б—В–≤–Є—О –ґ–Є—А–љ—Л—Е –Ї–Є—Б–ї–Њ—В [3].
–Ґ—А–Њ—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є —Б—В–∞—В—Г—Б. –Ъ–∞–Ї –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ—Л–є –љ—Г—В—А–Є—В–Є–≤–љ—Л–є —Б—В–∞—В—Г—Б, —В–∞–Ї –Є –Є–Ј–±—Л—В–Њ—З–љ—Л–є —П–≤–ї—П—О—В—Б—П —Д–∞–Ї—В–Њ—А–∞–Љ–Є —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є. –Т –Њ–і–љ–Њ–Љ –Є–Ј –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –њ–Њ—Б–≤—П—Й–µ–љ–љ—Л—Е —Н—В–Њ–є —В–µ–Љ–∞—В–Є–Ї–µ, –±—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–љ—Л–Љ –і–Є–∞–≥–љ–Њ–Ј–Њ–Љ (–∞–ї–Ї–Њ–≥–Њ–ї—М–љ–∞—П –±–Њ–ї–µ–Ј–љ—М –њ–µ—З–µ–љ–Є) –љ–Є–Ј–Ї–Є–є –Ї–∞–ї–Њ—А–∞–ґ –Ї–Њ—А—А–µ–ї–Є—А—Г–µ—В —Б –±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Њ–є —Б–Љ–µ—А—В–љ–Њ—Б—В—М—О –≤ —В–µ—З–µ–љ–Є–µ 6 –Љ–µ—Б—П—Ж–µ–≤. –Т –і—А—Г–≥–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ —Г –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤ —Б –Њ–ґ–Є—А–µ–љ–Є–µ–Љ —А–Є—Б–Ї –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є –≤ 2–3 —А–∞–Ј–∞ –≤—Л—И–µ –≤ —Б—А–∞–≤–љ–µ–љ–Є–Є —Б –ї–Є—Ж–∞–Љ–Є —Б –љ–Њ—А–Љ–∞–ї—М–љ–Њ–є –Љ–∞—Б—Б–Њ–є —В–µ–ї–∞. –Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –Њ–ґ–Є—А–µ–љ–Є–µ —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–µ—В—Б—П, –Ї–∞–Ї –љ–µ–Ј–∞–≤–Є—Б–Є–Љ—Л–є —Д–∞–Ї—В–Њ—А —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П —Б—В–µ–∞—В–Њ–Ј–∞ –Є —Б—В–µ–∞—В–Њ–≥–µ–њ–∞—В–Є—В–∞. –Т —Б–ї—Г—З–∞–µ, –Ї–Њ–≥–і–∞ –Ј–ї–Њ—Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ –∞–ї–Ї–Њ–≥–Њ–ї—П –љ–∞–Ї–ї–∞–і—Л–≤–∞–µ—В—Б—П –љ–∞ –Њ–ґ–Є—А–µ–љ–Є–µ, —А–Є—Б–Ї –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–µ—З–µ–љ–Є —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –њ—А–Є–Љ–µ—А–љ–Њ –≤ 6 —А–∞–Ј [9].
–Я–Њ–Љ–Є–Љ–Њ –ґ–Є—А–Њ–≤ –Є –Ї–∞–ї–Њ—А–Є–є, –љ–∞ —А–∞–Ј–≤–Є—В–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–µ—З–µ–љ–Є —Г –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤ –≤–ї–Є—П–µ—В —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ –ґ–µ–ї–µ–Ј–∞ –≤ —А–∞—Ж–Є–Њ–љ–µ –њ–Є—В–∞–љ–Є—П. –•—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ —Н—В–∞–љ–Њ–ї–∞ —Г—Б–Є–ї–Є–≤–∞–µ—В –∞–±—Б–Њ—А–±—Ж–Є—О –ґ–µ–ї–µ–Ј–∞ –Є–Ј –Ї–Є—И–µ—З–љ–Є–Ї–∞, –Є —З–µ—А–µ–Ј –љ–µ–Ї–Њ—В–Њ—А–Њ–µ –≤—А–µ–Љ—П —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –Ј–∞–њ–∞—Б –ґ–µ–ї–µ–Ј–∞ –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ. –Ц–µ–ї–µ–Ј–Њ – –Ї–∞—В–∞–ї–Є–Ј–∞—В–Њ—А –њ—А–Њ–і—Г–Ї—Ж–Є–Є —Б–≤–Њ–±–Њ–і–љ—Л—Е —А–∞–і–Є–Ї–∞–ї–Њ–≤ –Є –Љ–Њ–ґ–µ—В —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞—В—М –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О –њ–µ—З–µ–љ–Є, —Г—Б–Є–ї–Є–≤–∞—П –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л–є —Б—В—А–µ—Б—Б, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–є –і–ї—П –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ —Н—В–∞–љ–Њ–ї–∞ –≤ –њ–µ—З–µ–љ–Є [4].
–Т–Є—А—Г—Б–љ—Л–µ –≥–µ–њ–∞—В–Є—В—Л. –Я—А–Є–±–ї–Є–Ј–Є—В–µ–ї—М–љ–Њ –Њ—В 18 –і–Њ 25% –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤ –Є–љ—Д–Є—Ж–Є—А–Њ–≤–∞–љ—Л –≤–Є—А—Г—Б–Њ–Љ –≥–µ–њ–∞—В–Є—В–∞ –° (HCV). –£ –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤ —Б –±–Њ–ї–µ–Ј–љ—М—О –њ–µ—З–µ–љ–Є —З–∞—Б—В–Њ—В–∞ HCV –Є–љ—Д–µ–Ї—Ж–Є–Є –≤—Л—И–µ – –±–Њ–ї–µ–µ 40%. –°–Њ—З–µ—В–∞–љ–Є–µ –∞–ї–Ї–Њ–≥–Њ–ї—П —Б HCV –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Г—Б–Ї–Њ—А—П–µ—В –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–µ—З–µ–љ–Є, –њ—А–Є–±–ї–Є–Ј–Є—В–µ–ї—М–љ–Њ –≤ 8–10 —А–∞–Ј. –Ъ–∞–Ї –Є HCV, –≤–Є—А—Г—Б –≥–µ–њ–∞—В–Є—В–∞ –Т (HBV) —Г—Б–Ї–Њ—А—П–µ—В —В–µ–Љ–њ—Л —А–∞–Ј–≤–Є—В–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –≠–њ–Є–і–µ–Љ–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –і–∞–љ–љ—Л–µ –Њ—В—А–∞–ґ–∞—О—В –±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї—Г—О —Б–Љ–µ—А—В–љ–Њ—Б—В—М –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤ –≤ —Б–Њ—З–µ—В–∞–љ–Є–Є —Б HBV –Є–љ—Д–µ–Ї—Ж–Є–µ–є [10].
–Ф–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞
–Ъ–∞–Ї –њ—А–Є—З–Є–љ—Г –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є, –∞–ї–Ї–Њ–≥–Њ–ї—М —Б–ї–µ–і—Г–µ—В —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞—В—М —Г –ї–Є—Ж, —Г–њ–Њ—В—А–µ–±–ї—П—О—Й–Є—Е –±–Њ–ї–µ–µ 80 –≥ —Н—В–∞–љ–Њ–ї–∞ –≤ —Б—Г—В–Ї–Є. –Ю–і–љ–∞–Ї–Њ —Б–ї–µ–і—Г–µ—В —Г—З–Є—В—Л–≤–∞—В—М, —З—В–Њ –њ—А–Є –љ–∞–ї–Є—З–Є–Є –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞ (–љ–∞–њ—А–Є–Љ–µ—А, –њ–Њ–ї –Є HCV) –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–∞—П –±–Њ–ї–µ–Ј–љ—М –њ–µ—З–µ–љ–Є –Љ–Њ–ґ–µ—В –±—Л—В—М –і–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞–љ–∞ –Є –њ—А–Є –≥–Њ—А–∞–Ј–і–Њ –Љ–µ–љ—М—И–µ–Љ –Ї–Њ–ї–Є—З–µ—Б—В–≤–µ —Г–њ–Њ—В—А–µ–±–ї—П–µ–Љ–Њ–≥–Њ —Н—В–∞–љ–Њ–ї–∞. –°–ї–Њ–ґ–љ–Њ—Б—В–Є –≤ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–µ –Љ–Њ–≥—Г—В –±—Л—В—М —Б–≤—П–Ј–∞–љ—Л —Б —В–µ–Љ, —З—В–Њ —З–∞—Б—В—М –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б–Ї—А—Л–≤–∞–µ—В —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ –∞–ї–Ї–Њ–≥–Њ–ї—П. –Т —В–∞–Ї–Њ–Љ —Б–ї—Г—З–∞–µ –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ —Б–њ–µ—Ж–Є–∞–ї—М–љ—Л—Е –Њ–њ—А–Њ—Б–љ–Є–Ї–Њ–≤, –±–µ—Б–µ–і–∞ —Б —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–∞–Љ–Є –Є –Ј–љ–∞–Ї–Њ–Љ—Л–Љ–Є –±–Њ–ї—М–љ–Њ–≥–Њ –і–ї—П –њ–Њ—Б—В–∞–љ–Њ–≤–Ї–Є –і–Є–∞–≥–љ–Њ–Ј–∞.
–Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л–є —Д–Є–Ј–Є–Ї–∞–ї—М–љ—Л–є —Б–Є–Љ–њ—В–Њ–Љ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ—М—О –њ–µ—З–µ–љ–Є – —Н—В–Њ –≥–µ–њ–∞—В–Њ–Љ–µ–≥–∞–ї–Є—П. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –≤—Б—В—А–µ—З–∞—О—В—Б—П —Б–Є–Љ–њ—В–Њ–Љ—Л, –њ–µ—А–µ—З–Є—Б–ї–µ–љ–љ—Л–µ –≤ —В–∞–±–ї–Є—Ж–µ 1.
–Я—А–Є–±–ї–Є–Ј–Є—В–µ–ї—М–љ–Њ —Г 75% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ—М—О –њ–µ—З–µ–љ–Є –≤—Л—П–≤–ї—П–µ—В—Б—П –Љ–∞–Ї—А–Њ—Ж–Є—В–∞—А–љ–∞—П –∞–љ–µ–Љ–Є—П. –Ъ–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –Њ—В–Љ–µ—З–∞–µ—В—Б—П —Г–Љ–µ—А–µ–љ–љ—Л–є –ї–µ–є–Ї–Њ—Ж–Є—В–Њ–Ј, –њ–Њ–≤—Л—И–µ–љ–Є–µ –Р–Ы–Ґ –Є –Р–°–Ґ, —Г—А–Њ–≤–љ–Є –Ї–Њ—В–Њ—А—Л—Е —А–µ–і–Ї–Њ –њ—А–µ–≤—Л—И–∞—О—В 300 –µ–і/–ї –Є –љ–µ –Ї–Њ—А—А–µ–ї–Є—А—Г—О—В —Б —В—П–ґ–µ—Б—В—М—О –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –±–Њ–ї–µ–µ –Ј–љ–∞—З–Є–Љ—Л–Љ –Љ–Њ–ґ–µ—В –±—Л—В—М –њ–Њ–≤—Л—И–µ–љ–Є–µ –≥–∞–Љ–Љ–∞––≥–ї—Г—В–∞–Љ–Є–ї—В—А–∞–љ—Б–њ–µ–њ—В–Є–і–∞–Ј—Л (–У–У–Ґ–Я). –£—А–Њ–≤–µ–љ—М –±–Є–ї–Є—А—Г–±–Є–љ–∞ –≤ —Б—Л–≤–Њ—А–Њ—В–Ї–µ –Є –њ—А–Њ—В—А–Њ–Љ–±–Є–љ–Њ–≤–Њ–µ –≤—А–µ–Љ—П –Є—Б–њ–Њ–ї—М–Ј—Г—О—В—Б—П –≤ –Ї–∞—З–µ—Б—В–≤–µ –њ—А–µ–і–Є–Ї—В–Њ—А–Њ–≤ —В—П–ґ–µ—Б—В–Є –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–µ—З–µ–љ–Є. –Ь—Н–і–і—А–µ–є —Б —Б–Њ–∞–≤—В. –≤ 1978 –≥. –њ—А–µ–і–ї–Њ–ґ–Є–ї–Є –љ–∞ –Њ—Б–љ–Њ–≤–µ —Н—В–Є—Е –і–≤—Г—Е –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є —А–∞—Б—З–µ—В–љ—Л–є –Є–љ–і–µ–Ї—Б, –њ–Њ–ї—Г—З–Є–≤—И–Є–є –љ–∞–Ј–≤–∞–љ–Є–µ –Є–љ–і–µ–Ї—Б–∞ –Ь—Н–і–і—А–µ—П (–Ш–Ь):
–Ш–Ь = 4,6 —Е (–Я–Т –≤ —Б–µ–Ї—Г–љ–і–∞—Е – –Я–Т –Ї–Њ–љ—В—А–Њ–ї—М –≤ —Б–µ–Ї—Г–љ–і–∞—Е) + –±–Є–ї–Є—А—Г–±–Є–љ (–Љ–≥/–і–ї),
–µ—Б–ї–Є –Ш–Ь > 32, —В–Њ —Б–Љ–µ—А—В–љ–Њ—Б—В—М —Б–Њ—Б—В–∞–≤–ї—П–µ—В –Њ–Ї–Њ–ї–Њ 50% –≤ —В–µ—З–µ–љ–Є–µ 1 –Љ–µ—Б—П—Ж–∞.
–Я—А–Є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–µ —Б–ї–µ–і—Г–µ—В –њ–Њ–Љ–љ–Є—В—М, —З—В–Њ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –њ–µ—З–µ–љ–Є –њ–Њ–і –і–µ–є—Б—В–≤–Є–µ–Љ —Н—В–∞–љ–Њ–ї–∞ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –љ–∞ —Д–Њ–љ–µ –±–Њ–ї–µ–µ –Њ–±—И–Є—А–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П – –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є, –і–ї—П –Ї–Њ—В–Њ—А–Њ–є —Е–∞—А–∞–Ї—В–µ—А–љ–Њ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Љ–љ–Њ–≥–Є—Е –Њ—А–≥–∞–љ–Њ–≤ –Є —Б–Є—Б—В–µ–Љ (–∞–ї–Ї–Њ–≥–Њ–ї—М–љ—Л–є –њ–∞–љ–Ї—А–µ–∞—В–Є—В, –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є—П, –њ–Њ—А–∞–ґ–µ–љ–Є–µ —Ж–µ–љ—В—А–∞–ї—М–љ–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Є —В.–і.). –Ґ–∞–Ї–ґ–µ —А–∞–Ј—А–∞–±–Њ—В–∞–љ–Њ –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ —В–µ—Б—В–Њ–≤ –і–ї—П –Њ—Ж–µ–љ–Ї–Є –Љ–∞—А–Ї–µ—А–Њ–≤ —Г–њ–Њ—В—А–µ–±–ї–µ–љ–Є—П –∞–ї–Ї–Њ–≥–Њ–ї—П. –Ъ —В–∞–Ї–Є–Љ —В–µ—Б—В–∞–Љ –Њ—В–љ–Њ—Б—П—В—Б—П: –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–∞—П –Р–°–Ґ, –±–µ–Ј—Г–≥–ї–µ–≤–Њ–і–љ—Л–є —В—А–∞–љ—Б—Д–µ—А—А–Є–љ (–і–µ—Б–Є–∞–ї–Њ—В—А–∞–љ—Б—Д–µ—А—А–Є–љ) –Є –∞–љ—В–Є—В–µ–ї–∞ –њ—А–Њ—В–Є–≤ –∞—Ж–µ—В–∞–ї—М–і–µ–≥–Є–і––±–µ–ї–Ї–Њ–≤—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤ [6].
–У–Є—Б—В–Њ–ї–Њ–≥–Є—П. –Ъ–ї–Є–љ–Є–Ї–Њ––Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є—П –њ–Њ–і—А–∞–Ј–і–µ–ї—П–µ—В –∞–ї–Ї–Њ–≥–Њ–ї—М–љ—Г—О –±–Њ–ї–µ–Ј–љ—М –њ–µ—З–µ–љ–Є –љ–∞ —В—А–Є –Њ—Б–љ–Њ–≤–љ—Л–µ —Д–Њ—А–Љ—Л: —Б—В–µ–∞—В–Њ–Ј, –≥–µ–њ–∞—В–Є—В –Є —Ж–Є—А—А–Њ–Ј. –Э–∞–Є–±–Њ–ї–µ–µ —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є: –ґ–Є—А–Њ–≤–∞—П –і–Є—Б—В—А–Њ—Д–Є—П –≥–µ–њ–∞—В–Њ—Ж–Є—В–Њ–≤, –љ–∞–ї–Є—З–Є–µ —В–µ–ї–µ—Ж –Ь—Н–ї–ї–Њ—А–Є, –љ–µ–є—В—А–Њ—Д–Є–ї—М–љ–∞—П –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є—П –Є –њ–µ—А–Є–≤–µ–љ—Г–ї—П—А–љ—Л–є —Д–Є–±—А–Њ–Ј. –°—В–µ–∞—В–Њ–Ј –≤—Б—В—А–µ—З–∞–µ—В—Б—П —Г 60–95% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ—М—О. –Ц–Є—А, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –ї–Њ–Ї–∞–ї–Є–Ј—Г–µ—В—Б—П –њ–µ—А–Є—Ж–µ–љ—В—А–∞–ї—М–љ–Њ, —Е–Њ—В—П –≤ —В—П–ґ–µ–ї—Л—Е —Б–ї—Г—З–∞—П—Е –Є–Љ–µ–µ—В –њ–∞–љ–ї–Њ–±—Г–ї—П—А–љ–Њ–µ —А–∞—Б–њ—А–µ–і–µ–ї–µ–љ–Є–µ. –С–Њ–ї–µ–µ —Е–∞—А–∞–Ї—В–µ—А–љ—Л–Љ —П–≤–ї—П–µ—В—Б—П –Љ–∞–Ї—А–Њ–≤–µ–Ј–Є–Ї—Г–ї—П—А–љ—Л–є —Б—В–µ–∞—В–Њ–Ј, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–Љ –ґ–Є—А–Њ–≤—Л–µ –≤–Ї–ї—О—З–µ–љ–Є—П –≤ –≥–µ–њ–∞—В–Њ—Ж–Є—В–∞—Е —Б–Љ–µ—Й–∞—О—В —П–і—А–Њ –Ї –њ–µ—А–Є—Д–µ—А–Є–Є –Ї–ї–µ—В–Ї–Є. –Ь–Є–Ї—А–Њ–≤–µ–Ј–Є–Ї—Г–ї—П—А–љ—Л–є —Б—В–µ–∞—В–Њ–Ј —В–∞–Ї–ґ–µ –≤—Б—В—А–µ—З–∞–µ—В—Б—П —Г –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤. –С–∞–ї–ї–Њ–љ–љ–∞—П –і–µ–≥–µ–љ–µ—А–∞—Ж–Є—П –≥–µ–њ–∞—В–Њ—Ж–Є—В–Њ–≤ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П —П–≤–љ—Л–Љ –Њ—В–µ–Ї–Њ–Љ –Ї–ї–µ—В–Ї–Є —Б –±–ї–µ–і–љ–Њ–є —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–Њ–є. –С–∞–ї–ї–Њ–љ–љ–∞—П –і–µ–≥–µ–љ–µ—А–∞—Ж–Є—П – –љ–µ—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–є –Љ–∞—А–Ї–µ—А –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –≥–µ–њ–∞—В–Њ—Ж–Є—В–Њ–≤, –Ї–Њ—В–Њ—А—Л–є —З–∞—Б—В–Њ —Б–Њ—З–µ—В–∞–µ—В—Б—П —Б –љ–∞–ї–Є—З–Є–µ–Љ –∞—Ж–Є–і–Њ—Д–Є–ї—М–љ—Л—Е (–∞–њ–Њ–њ—В–Њ—В–Є—З–µ—Б–Ї–Є—Е) —В–µ–ї–µ—Ж. –≠—В–Є –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –≤—Б—В—А–µ—З–∞—О—В—Б—П —Г 60–90% –њ–∞—Ж–Є–µ–љ—В–Њ–≤. –Ґ–∞–Ї–ґ–µ –і–Њ—Б—В–∞—В–Њ—З–љ–Њ —З–∞—Б—В–Њ (70–75%) —Г –љ–Є—Е –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П —В–µ–ї—М—Ж–∞ –Ь—Н–ї–ї–Њ—А–Є: –њ–Њ–ї—Г–Ї—А—Г–≥–ї–Њ–є —Д–Њ—А–Љ—Л —Н–Њ–Ј–Є–љ–Њ—Д–Є–ї—М–љ—Л–µ —Б—В—А—Г–Ї—В—Г—А—Л, –њ—А–µ–і—Б—В–∞–≤–ї—П—О—Й–Є–µ —Б–Њ–±–Њ–є –њ—А–Њ–Љ–µ–ґ—Г—В–Њ—З–љ—Л–µ —Д–Є–ї–∞–Љ–µ–љ—В—Л, –њ–Њ–і–≤–µ—А–≥—И–Є–µ—Б—П –Ї–Њ–љ–і–µ–љ—Б–∞—Ж–Є–Є. –Э–µ—Б–Љ–Њ—В—А—П –љ–∞ –Є—Е –љ–∞–ї–Є—З–Є–µ —Г –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–≥–Њ —З–Є—Б–ї–∞ –±–Њ–ї—М–љ—Л—Е, —В–µ–ї—М—Ж–∞ –Ь—Н–ї–ї–Њ—А–Є –љ–µ –њ–∞—В–Њ–≥–љ–Њ–Љ–Њ–љ–Є—З–љ—Л –і–ї—П –∞–ї–Ї–Њ–≥–Њ–≥–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є. –Ю–љ–Є –≤—Б—В—А–µ—З–∞—О—В—Б—П —Г –±–Њ–ї—М–љ—Л—Е –љ–µ–∞–ї–Ї–Њ–≥–Њ–ї—М–љ—Л–Љ —Б—В–µ–∞—В–Њ–≥–µ–њ–∞—В–Є—В–Њ–Љ, –њ–µ—А–≤–Є—З–љ—Л–Љ –±–Є–ї–Є–∞—А–љ—Л–Љ —Ж–Є—А—А–Њ–Ј–Њ–Љ, –±–Њ–ї–µ–Ј–љ–Є –Т–Є–ї—М—Б–Њ–љ–∞, –∞ —В–∞–Ї–ґ–µ –љ–∞ —Д–Њ–љ–µ –њ—А–Є–µ–Љ–∞ –≥—А–Є–Ј–µ–Њ—Д—Г–ї—М–≤–Є–љ–∞ –Є –∞–Љ–Є–Њ–і–∞—А–Њ–љ–∞. –Т–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ–µ—З–µ–љ–Є –љ–∞–±–ї—О–і–∞—О—В—Б—П —Г 50–85% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ—М—О –њ–µ—З–µ–љ–Є. –Я–Њ–ї–Є–Љ–Њ—А—Д–љ–Њ—П–і–µ—А–љ—Л–µ –ї–µ–є–Ї–Њ—Ж–Є—В—Л (–љ–µ–є—В—А–Њ—Д–Є–ї—Л) –њ—А–µ–Њ–±–ї–∞–і–∞—О—В –≤ –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–Љ –Є–љ—Д–Є–ї—М—В—А–∞—В–µ, —З—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –Њ—В–ї–Є—З–Є—В—М –∞–ї–Ї–Њ–≥–Њ–ї—М––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Њ—В –і—А—Г–≥–Є—Е –њ—А–Є—З–Є–љ, –≤—Л–Ј—Л–≤–∞—О—Й–Є—Е –≥–µ–њ–∞—В–Є—В. –Ґ–∞–Ї–ґ–µ –≤—Б—В—А–µ—З–∞—О—В—Б—П –Љ–Њ–љ–Њ–љ—Г–Ї–ї–µ–∞—А–љ—Л–µ –Ї–ї–µ—В–Ї–Є. –§–Є–±—А–Њ–Ј –њ—А–Є –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–Є –њ–µ—З–µ–љ–Є –≤—Л—П–≤–ї—П–µ—В—Б—П —Г 50–75% –њ–∞—Ж–Є–µ–љ—В–Њ–≤. –Ю–љ –љ–∞—З–Є–љ–∞–µ—В—Б—П —Б –Њ—В–ї–Њ–ґ–µ–љ–Є–є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –≤–Њ–Ї—А—Г–≥ —В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є –њ–µ—З–µ–љ–Њ—З–љ–Њ–є –≤–µ–љ—Г–ї—Л –Є —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ—П–µ—В—Б—П –≤ –њ–µ—З–µ–љ–Њ—З–љ—Г—О –њ–∞—А–µ–љ—Е–Є–Љ—Г. –Я–Њ –Љ–µ—А–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –Њ–±—А–∞–Ј—Г—О—В—Б—П —Ж–µ–љ—В—А–Њ–—Ж–µ–љ—В—А–∞–ї—М–љ—Л–µ –Є —Ж–µ–љ—В—А–Њ––њ–Њ—А—В–∞–ї—М–љ—Л–µ —Б–µ–њ—В—Л. –¶–Є—А—А–Њ–Ј –њ–µ—З–µ–љ–Є, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –Љ–Є–Ї—А–Њ–љ–Њ–і—Г–ї—П—А–љ—Л–є [2–4].
–Ф–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ—Л–є –і–Є–∞–≥–љ–Њ–Ј. –Ю–њ—А–µ–і–µ–ї–µ–љ–љ—Л–µ —Б–ї–Њ–ґ–љ–Њ—Б—В–Є –≤–Њ–Ј–љ–Є–Ї–∞—О—В –њ—А–Є –і–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ–Њ–є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–µ –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є –Є –љ–µ–∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–≥–Њ —Б—В–µ–∞—В–Њ–≥–µ–њ–∞—В–Є—В–∞ (–Э–Р–°–У). –Ю–±–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Є–Љ–µ—О—В —Б—Е–Њ–і–љ—Г—О –Ї–ї–Є–љ–Є—З–µ—Б–Ї—Г—О –Ї–∞—А—В–Є–љ—Г –Є –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Є. –Я–Њ–Љ–Њ—Й—М –Љ–Њ–ґ–µ—В –Њ–Ї–∞–Ј–∞—В—М –Њ—Ж–µ–љ–Ї–∞ —Б–Њ–Њ—В–љ–Њ—И–µ–љ–Є—П –Р–°–Ґ/–Р–Ы–Ґ. –£ –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤ —Н—В–Њ —Б–Њ–Њ—В–љ–Њ—И–µ–љ–Є–µ, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –њ—А–µ–≤—Л—И–∞–µ—В 2, –∞ –њ—А–Є –Э–Р–°–У —Б—А–µ–і–љ–µ–µ –Ј–љ–∞—З–µ–љ–Є–µ —Н—В–Њ–≥–Њ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї—П —Б–Њ—Б—В–∞–≤–ї—П–µ—В 1,0. –Я–Њ –Љ–µ—А–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П —Д–Є–±—А–Њ–Ј–∞ –њ—А–Є –Э–Р–°–У —Н—В–Њ —Б–Њ–Њ—В–љ–Њ—И–µ–љ–Є–µ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П, –Њ–і–љ–∞–Ї–Њ –і–∞–ґ–µ –љ–∞ –њ–Њ–Ј–і–љ–Є—Е —Б—В–∞–і–Є—П—Е —А–µ–і–Ї–Њ –њ—А–µ–≤—Л—И–∞–µ—В 1,5.
–Ю—Б–ї–Њ–ґ–љ–µ–љ–Є—П. –Ю—Б–ї–Њ–ґ–љ–µ–љ–Є—П –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є —В–Є–њ–Є—З–љ—Л –і–ї—П –ї—О–±–Њ–≥–Њ —В–Є–њ–∞ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–µ—З–µ–љ–Є. –≠—В–Њ –Є –∞—Б—Ж–Є—В, –Є –ґ–µ–ї—Г–і–Њ—З–љ–Њ––Ї–Є—И–µ—З–љ–Њ–µ –Ї—А–Њ–≤–Њ—В–µ—З–µ–љ–Є–µ, –Є –≥–Є–њ–Њ–њ—А–Њ—В—А–Њ–Љ–±–Є–љ–µ–Љ–Є—П –Є —В.–і. –Я—А–Є —А–∞–Ј–≤–Є—В–Є–Є –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–≥–Њ —Ж–Є—А—А–Њ–Ј–∞ –Љ–Њ–ґ–µ—В —А–∞–Ј–≤–Є–≤–∞—В—М—Б—П –≥–µ–њ–∞—В–Њ—Ж–µ–ї–ї—О–ї—П—А–љ–∞—П –Ї–∞—А—Ж–Є–љ–Њ–Љ–∞. –Э–Њ –Є —Б–∞–Љ –∞–ї–Ї–Њ–≥–Њ–ї—М —П–≤–ї—П–µ—В—Б—П –љ–µ–Ј–∞–≤–Є—Б–Є–Љ—Л–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ —А–∞–Ї–∞ –њ–µ—З–µ–љ–Є, –Њ–і–љ–∞–Ї–Њ –≤ –Љ–µ–љ—М—И–µ–є —Б—В–µ–њ–µ–љ–Є, —З–µ–Љ –≤–Є—А—Г—Б—Л –≥–µ–њ–∞—В–Є—В–∞. –Я–Њ—В—А–µ–±–ї–µ–љ–Є–µ –∞–ї–Ї–Њ–≥–Њ–ї—П –љ–∞ —Д–Њ–љ–µ HBV –Є–љ—Д–µ–Ї—Ж–Є–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –њ–Њ–≤—Л—И–∞–µ—В —А–Є—Б–Ї —А–∞–Ј–≤–Є—В–Є—П –≥–µ–њ–∞—В–Њ—Ж–µ–ї–ї—О–ї—П—А–љ–Њ–є –Ї–∞—А—Ж–Є–љ–Њ–Љ—Л. –°—А–µ–і–Є –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ї–Њ–≤ –љ–∞–Є–±–Њ–ї–µ–µ —Г—П–Ј–≤–Є–Љ–Њ–є –≥—А—Г–њ–њ–Њ–є –≤ –Њ—В–љ–Њ—И–µ–љ–Є–Є —А–∞–Ј–≤–Є—В–Є—П —А–∞–Ї–∞ –њ–µ—З–µ–љ–Є —П–≤–ї—П—О—В—Б—П –Љ—Г–ґ—З–Є–љ—Л —Б—В–∞—А—И–µ 50 –ї–µ—В [2].
–Ы–µ—З–µ–љ–Є–µ
–Р–±—Б—В–Є–љ–µ–љ—Ж–Є—П. –Ю—Б–љ–Њ–≤–∞ –ї–µ—З–µ–љ–Є—П –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є – –њ—А–µ–Ї—А–∞—Й–µ–љ–Є–µ –њ—А–Є–µ–Љ–∞ –∞–ї–Ї–Њ–≥–Њ–ї—П. –Р–±—Б—В–Є–љ–µ–љ—Ж–Є—П —Г–ї—Г—З—И–∞–µ—В –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В–Є –і–∞–ґ–µ –њ—А–Є —Ж–Є—А—А–Њ–Ј–µ –њ–µ—З–µ–љ–Є –Є –њ–Њ—А—В–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є [4].
–Ъ–Њ—А—А–µ–Ї—Ж–Є—П —В—А–Њ—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ —Б—В–∞—В—Г—Б–∞. –Т–∞–ґ–љ—Л–Љ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–Љ –ї–µ—З–µ–љ–Є—П —П–≤–ї—П–µ—В—Б—П –∞–і–µ–Ї–≤–∞—В–љ–Њ–µ –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є–µ –њ–Є—В–∞—В–µ–ї—М–љ—Л—Е –≤–µ—Й–µ—Б—В–≤. –≠–љ–µ—А–≥–µ—В–Є—З–µ—Б–Ї–∞—П —Ж–µ–љ–љ–Њ—Б—В—М –і–Є–µ—В—Л –і–Њ–ї–ґ–љ–∞ –±—Л—В—М –љ–µ –Љ–µ–љ–µ–µ 2000 –Ї–∞–ї–Њ—А–Є–є –≤ —Б—Г—В–Ї–Є —Б —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ–Љ –±–µ–ї–Ї–∞ 1 –≥ –љ–∞ 1 –Ї–≥ –Љ–∞—Б—Б—Л —В–µ–ї–∞ –Є –і–Њ—Б—В–∞—В–Њ—З–љ—Л–Љ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ–Љ –≤–Є—В–∞–Љ–Є–љ–Њ–≤ [9].
–У–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ—Б—В–µ—А–Њ–Є–і—Л. –Т–Њ–њ—А–Њ—Б –Њ –љ–∞–Ј–љ–∞—З–µ–љ–Є–Є –Ї–Њ—А—В–Є–Ї–Њ—Б—В–µ—А–Њ–Є–і–љ–Њ–є —В–µ—А–∞–њ–Є–Є –Њ—Б—В–∞–µ—В—Б—П –љ–µ–Њ–і–љ–Њ–Ј–љ–∞—З–љ—Л–Љ. –†–µ–Ј—Г–ї—М—В–∞—В—Л 13 —А–∞–љ–і–Њ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е –Ї–Њ–љ—В—А–Њ–ї–Є—А–Њ–≤–∞–љ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є —Г–Ї–∞–Ј—Л–≤–∞—О—В –љ–∞ –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ–є –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В–Є —В–∞–Ї–Є—Е –±–Њ–ї—М–љ—Л—Е. –Я–Њ–Ї–∞–Ј–∞–љ–Є–µ–Љ –Ї –љ–∞–Ј–љ–∞—З–µ–љ–Є—О —Б—В–µ—А–Њ–Є–і–Њ–≤ —П–≤–ї—П–µ—В—Б—П –Є–љ–і–µ–Ї—Б –Ь—Н–і–і—А–µ—П > 32, —З—В–Њ, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–Њ —Б —В—П–ґ–µ–ї—Л–Љ –∞–ї–Ї–Њ–≥–Њ–ї—М–љ—Л–Љ –≥–µ–њ–∞—В–Є—В–Њ–Љ. –Ю–±—Л—З–љ–Њ —Н—Д—Д–µ–Ї—В–Є–≤–љ–∞—П –і–Њ–Ј–∞ —Н—В–Њ 40 –Љ–≥ –њ—А–µ–і–љ–Є–Ј–Њ–ї–Њ–љ–∞ –≤ —Б—Г—В–Ї–Є (–Є–ї–Є 32 –Љ–≥ –Љ–µ—В–Є–ї–њ—А–µ–і–љ–Є–Ј–Њ–ї–Њ–љ–∞). –Я—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ—Б—В—М –ї–µ—З–µ–љ–Є—П –≤ —Б—А–µ–і–љ–µ–Љ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 28–30 –і–љ–µ–є. –Ц–µ–ї—Г–і–Њ—З–љ–Њ––Ї–Є—И–µ—З–љ—Л–µ –Ї—А–Њ–≤–Њ—В–µ—З–µ–љ–Є—П –Є –њ–Њ—З–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М –Њ–≥—А–∞–љ–Є—З–Є–≤–∞—О—В –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –њ—А–Є–Љ–µ–љ–µ–љ–Є—П –≥–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ—Б—В–µ—А–Њ–Є–і–Њ–≤, –Њ–і–љ–∞–Ї–Њ –Є—Е –љ–∞–Ј–љ–∞—З–µ–љ–Є–µ –љ–µ —Б–Њ–њ—А—П–ґ–µ–љ–Њ —Б –њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є. –Ю—В–љ–Њ—Б–Є—В–µ–ї—М–љ—Л–Љ–Є –њ—А–Њ—В–Є–≤–Њ–њ–Њ–Ї–∞–Ј–∞–љ–Є—П–Љ–Є –Ї –љ–∞–Ј–љ–∞—З–µ–љ–Є—О —Б—В–µ—А–Њ–Є–і–Њ–≤ —П–≤–ї—П—О—В—Б—П –∞–Ї—В–Є–≤–љ—Л–µ –Є–љ—Д–µ–Ї—Ж–Є–Є, –њ–∞–љ–Ї—А–µ–∞—В–Є—В, –Є–љ—Б—Г–ї–Є–љ–Њ–Ј–∞–≤–Є—Б–Є–Љ—Л–є —Б–∞—Е–∞—А–љ—Л–є –і–Є–∞–±–µ—В [2,9].
–Я–µ–љ—В–Њ–Ї—Б–Є—Д–Є–ї–ї–Є–љ. –Я–µ–љ—В–Њ–Ї—Б–Є—Д–Є–ї–ї–Є–љ —Г–Љ–µ–љ—М—И–∞–µ—В –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –Ј–∞ —Б—З–µ—В –њ–Њ–і–∞–≤–ї–µ–љ–Є—П —Б–Є–љ—В–µ–Ј–∞ –§–Э–Ю–α. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –±—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –Њ–љ –Њ–±–ї–∞–і–∞–µ—В –њ—А–Њ—Д–Є–ї–∞–Ї—В–Є—З–µ—Б–Ї–Є–Љ –і–µ–є—Б—В–≤–Є–µ–Љ –≤ –Њ—В–љ–Њ—И–µ–љ–Є–Є —А–∞–Ј–≤–Є—В–Є—П –≥–µ–њ–∞—В–Њ—А–µ–љ–∞–ї—М–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞. –Т–Њ–Ј–Љ–Њ–ґ–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ – —Н—В–Њ —Г–ї—Г—З—И–µ–љ–Є–µ –Љ–Є–Ї—А–Њ—Ж–Є—А–Ї—Г–ї—П—Ж–Є–Є (–≤ —З–∞—Б—В–љ–Њ—Б—В–Є, –≤ –њ–Њ—З–Ї–∞—Е) [3,11].
–≠—Б—Б–µ–љ—Ж–Є–∞–ї—М–љ—Л–µ —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і—Л. –Я–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –Љ–µ–Љ–±—А–∞–љ–љ—Л—Е —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і–Њ–≤ –≤–њ–µ—А–≤—Л–µ –±—Л–ї–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л —Г –ґ–Є–≤–Њ—В–љ—Л—Е, —Г –Ї–Њ—В–Њ—А—Л—Е –≤–Њ—Б–њ—А–Њ–Є–Ј–≤–Њ–і–Є–ї–Є –Љ–Њ–і–µ–ї—М –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є —З–µ–ї–Њ–≤–µ–Ї–∞. –Т —Б–≤—П–Ј–Є —Б —Н—В–Є–Љ —Н—Б—Б–µ–љ—Ж–Є–∞–ї—М–љ—Л–µ —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і—Л –љ–∞—И–ї–Є —Б–≤–Њ–µ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –њ—А–Є –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є. –Я—А–Є–Љ–µ—А–Њ–Љ —В–∞–Ї–Њ–≥–Њ –њ—А–µ–њ–∞—А–∞—В–∞ —П–≤–ї—П–µ—В—Б—П –≠—Б—Б–ї–Є–≤–µ—А® —Д–Њ—А—В–µ, –≤ —Б–Њ—Б—В–∞–≤ –Ї–Њ—В–Њ—А–Њ–≥–Њ, –њ–Њ–Љ–Є–Љ–Њ —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і–Њ–≤, –≤—Е–Њ–і—П—В –≤–Є—В–∞–Љ–Є–љ—Л –≥—А—Г–њ–њ—Л –Т –Є –Х. –Я—А–µ–њ–∞—А–∞—В –љ–Њ—А–Љ–∞–ї–Є–Ј—Г–µ—В –±–Є–Њ—Б–Є–љ—В–µ–Ј —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і–Њ–≤ –њ—А–Є –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є. –Т–Њ—Б—Б—В–∞–љ–∞–≤–ї–Є–≤–∞–µ—В –Љ–µ–Љ–±—А–∞–љ—Л –≥–µ–њ–∞—В–Њ—Ж–Є—В–Њ–≤ –њ—Г—В–µ–Љ —Б—В—А—Г–Ї—В—Г—А–љ–Њ–є —А–µ–њ–∞—А–∞—Ж–Є–Є –Є –Ј–∞ —Б—З–µ—В –Ї–Њ–љ–Ї—Г—А–µ–љ—В–љ–Њ–≥–Њ –Є–љ–≥–Є–±–Є—А–Њ–≤–∞–љ–Є—П –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤; —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —А–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –Ї–ї–µ—В–Њ–Ї –њ–µ—З–µ–љ–Є –Є —Б—В–∞–±–Є–ї–Є–Ј–Є—А—Г–µ—В —Д–Є–Ј–Є–Ї–Њ–—Е–Є–Љ–Є—З–µ—Б–Ї–Є–µ —Б–≤–Њ–є—Б—В–≤–∞ –ґ–µ–ї—З–Є. –≠—Б—Б–ї–Є–≤–µ—А® —Д–Њ—А—В–µ –љ–∞–Ј–љ–∞—З–∞—О—В –њ–Њ 2 –Ї–∞–њ—Б—Г–ї—Л 2–3 —А–∞–Ј–∞ –≤ —Б—Г—В–Ї–Є. –Т —А—П–і–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –±—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ —Н—Б—Б–µ–љ—Ж–Є–∞–ї—М–љ—Л–µ —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і—Л –Љ–Њ–≥—Г—В —Г–Љ–µ–љ—М—И–∞—В—М —В–µ–Љ–њ—Л —А–∞–Ј–≤–Є—В–Є—П —Д–Є–±—А–Њ–Ј–∞ –≤ –њ–µ—З–µ–љ–Є. –Ґ–∞–Ї, –≤ –Ї—Г–ї—М—В—Г—А–µ –Ї–ї–µ—В–Њ–Ї –Њ–љ–Є —Б–њ–Њ—Б–Њ–±–љ—Л –њ–Њ–і–∞–≤–ї—П—В—М –∞–Ї—В–Є–≤–∞—Ж–Є—О –Ј–≤–µ–Ј–і—З–∞—В—Л—Е –Ї–ї–µ—В–Њ–Ї. –Ъ—А–Њ–Љ–µ —Н—В–Њ–≥–Њ, –Њ–љ–Є —Г–Љ–µ–љ—М—И–∞—О—В –Є–љ–і—Г–Ї—Ж–Є—О —Ж–Є—В–Њ—Е—А–Њ–Љ–∞ 2–Х1 –Є –≤—Л–Ј–≤–∞–љ–љ—Л–є —Н—В–∞–љ–Њ–ї–Њ–Љ –∞–њ–Њ–њ—В–Њ–Ј –Ї–ї–µ—В–Њ–Ї in vivo, —Г–Љ–µ–љ—М—И–∞—О—В –њ–µ—А–µ–Ї–Є—Б–љ–Њ–µ –Њ–Ї–Є—Б–ї–µ–љ–Є–µ –ї–Є–њ–Є–і–Њ–≤ –Є –≤–Њ—Б—Б—В–∞–љ–∞–≤–ї–Є–≤–∞—О—В –≥–ї—Г—В–∞—В–Є–Њ–љ –≤ –њ–µ—З–µ–љ–Є [3].
S––∞–і–µ–љ–Њ–Ј–Є–ї–L––Љ–µ—В–Є–Њ–љ–Є–љ (–∞–і–µ–Љ–µ—В–Є–Њ–љ–Є–љ). –Р–і–µ–Љ–µ—В–Є–Њ–љ–Є–љ – —Н—В–Њ –њ—А–Є—А–Њ–і–љ–Њ–µ –≤–µ—Й–µ—Б—В–≤–Њ, –Ї–Њ—В–Њ—А–Њ–µ –Њ–±—А–∞–Ј—Г–µ—В—Б—П –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ –Є–Ј –Љ–µ—В–Є–Њ–љ–Є–љ–∞ —Б –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ –Р–Ґ–§ –њ—А–Є —Г—З–∞—Б—В–Є–Є —Д–µ—А–Љ–µ–љ—В–∞ S––∞–і–µ–љ–Њ–Ј–Є–ї–Љ–µ—В–Є–Њ–љ–Є–љ—Б–Є–љ—В–µ—В–∞–Ј—Л. –Р–і–µ–Љ–µ—В–Є–Њ–љ–Є–љ –≤—Е–Њ–і–Є—В –≤ —Б–Њ—Б—В–∞–≤ —А–∞–Ј–ї–Є—З–љ—Л—Е —В–Ї–∞–љ–µ–є –Њ—А–≥–∞–љ–Є–Ј–Љ–∞ –Є –Є–≥—А–∞–µ—В –≤–∞–ґ–љ—Г—О —А–Њ–ї—М –≤ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–µ –љ—Г–Ї–ї–µ–Є–љ–Њ–≤—Л—Е –Ї–Є—Б–ї–Њ—В, —Г–ї—Г—З—И–∞–µ—В —Д—Г–љ–Ї—Ж–Є—О –≥–µ–њ–∞—В–Њ—Ж–Є—В–Њ–≤ –Ј–∞ —Б—З–µ—В —Г–≤–µ–ї–Є—З–µ–љ–Є—П —В–µ–Ї—Г—З–µ—Б—В–Є –Љ–µ–Љ–±—А–∞–љ —З–µ—А–µ–Ј —А–µ–∞–Ї—Ж–Є—О —В—А–∞–љ—Б–Љ–µ—В–Є–ї–Є—А–Њ–≤–∞–љ–Є—П –Є –њ–Њ–≤—Л—И–∞–µ—В –њ—А–Њ–і—Г–Ї—Ж–Є—О –∞–љ—В–Є–Њ–Ї—Б–Є–і–∞–љ—В–Њ–≤. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ —В–Њ–Љ, —З—В–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ —Н—В–Њ–≥–Њ –њ—А–µ–њ–∞—А–∞—В–∞ –≤ –ї–µ—З–µ–љ–Є–Є –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є –њ–Њ–≤—Л—И–∞–µ—В —Г—А–Њ–≤–µ–љ—М –≥–ї—Г—В–∞—В–Є–Њ–љ–∞ –≤ –њ–µ—З–µ–љ–Є, –∞ —В–∞–Ї–ґ–µ –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ –≤–ї–Є—П–µ—В –љ–∞ –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М —Н—В–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ [8].
–Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –≤ –ї–Є—В–µ—А–∞—В—Г—А–µ –Њ–±—Б—Г–ґ–і–∞–µ—В—Б—П –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —Е–Є–Љ–µ—А–љ—Л—Е –∞–љ—В–Є—В–µ–ї –Ї –§–Э–Ю–α (–Є–љ—Д–ї–Є–Ї—Б–Є–Љ–∞–±), —Н—В–∞–љ–µ—А—Б–µ–њ—В (–±–ї–Њ–Ї–Є—А—Г–µ—В —А–∞—Б—В–≤–Њ—А–Є–Љ—Л–є –§–Э–Ю–α). –Ю–і–љ–∞–Ї–Њ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –Є—Е –Њ–≥—А–∞–љ–Є—З–µ–љ–Њ —А–∞–Љ–Ї–∞–Љ–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –Є –њ–Њ–±–Њ—З–љ—Л–Љ–Є —Н—Д—Д–µ–Ї—В–∞–Љ–Є —В–∞–Ї–Њ–є —В–µ—А–∞–њ–Є–Є.
–Ґ—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є—П –њ–µ—З–µ–љ–Є. –Ґ—А–∞–љ—Б–њ–ї–∞–љ—В–∞—Ж–Є—П –њ–µ—З–µ–љ–Є –њ–Њ–Ї–∞–Ј–∞–љ–∞ –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Б —Ж–Є—А—А–Њ–Ј–Њ–Љ –њ–µ—З–µ–љ–Є –Ї–ї–∞—Б—Б–∞ –° –њ–Њ –І–∞–є–ї–і––Я—М—О. –£—Б–ї–Њ–≤–Є–µ–Љ –њ–µ—А–µ—Б–∞–і–Ї–Є —П–≤–ї—П–µ—В—Б—П –∞–±—Б—В–Є–љ–µ–љ—Ж–Є—П, –Ї–∞–Ї –Љ–Є–љ–Є–Љ—Г–Љ, –≤ —В–µ—З–µ–љ–Є–µ –њ–Њ–ї—Г–≥–Њ–і–∞ –і–Њ –Њ–њ–µ—А–∞—Ж–Є–Є [12].
![–Ґ–∞–±–ї–Є—Ж–∞ 1. –І–∞—Б—В–Њ—В–∞ —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ –њ—А–Є –∞–ї–Ї–Њ–≥–Њ–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–µ—З–µ–љ–Є —Б—А–µ–і–Є –≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–Є—А—Г–µ–Љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ [3]](/data/articles/Image/t19/n17/1067-2.gif)
–Ы–Є—В–µ—А–∞—В—Г—А–∞
1. Kim W.R., Brown R.S., Terrault N.A., El–Serag H. Burden of liver disease in the United States: summary of a workship. Hepatology, 2002, 36, 227–242.
2. O’Shea R.S., Dasarathy S., McCullough A.J. Alcoholic liver disease. Hepatology, 2010, 51, 307–328.
3. Feldman M., Friedman L.S., Sleisenger M.H. Gastrointestinal and liver disease. Saunders, 2002, 1375–1391.
4. Kono H., Rusyn I., Yin M. NADPH oxidase–derived free radicals are key oxidants in alcohol–induced liver disease. J Clin Invest, 2000, 106, 867.
5. Kono H., Bradford B.U., Yin M. CYP2E1 is not involved in early alcohol–induced liver injury. Am J Physiol, 1999, 277, 1259.
6. Rolla R., Vay D., Mottaran E. Detection of circulating antibodies against malondialdehyde–acetaldehyde adducts in patients with alcohol–induced liver disease. Hepatology, 2000, 31, 878.
7. Casini A., Pinzani M., Milani S. Regulation of extracellular matrix syntesis by transforming growth factor beta 1 in human fat–storing cells. Gastroenterology, 1993, 105, 245.
8. –Р–±–і—Г—А–∞—Е–Љ–∞–љ–Њ–≤ –Ф.–Ґ. –Р–ї–Ї–Њ–≥–Њ–ї—М–љ—Л–є –≥–µ–њ–∞—В–Є—В. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –≥–µ–њ–∞—В–Њ–ї–Њ–≥–Є—П, 2008, 4(2), 3–10.
9. –С—Г–µ–≤–µ—А–Њ–≤ –Р.–Ю., –Я–∞–≤–ї–Њ–≤ –Р.–Ш., –Ш–≤–∞—И–Ї–Є–љ –Т.–Ґ. –Р–ї–Ї–Њ–≥–Њ–ї—М–љ–∞—П –±–Њ–ї–µ–Ј–љ—М –њ–µ—З–µ–љ–Є: –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ –ї–Є —Г–ї—Г—З—И–µ–љ–Є–µ –њ—А–Њ–≥–љ–Њ–Ј–∞? –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ–µ—А—Б–њ–µ–Ї—В–Є–≤—Л –≥–∞—Б—В—А–Њ—Н–љ—В–µ—А–Њ–ї–Њ–≥–Є–Є –Є –≥–µ–њ–∞—В–Њ–ї–Њ–≥–Є–Є, 2011, 2, 3–10.
10. Wheeler M. Ethanol and HCV–induced cytotoxicity: the perfect storm. Gastroenterology, 2005, 128, 232–234.
11. Akriviadis E., Bolta R., Briggs W. Pentoxifylline improves short–term survival in severe acute alcoholic hepatitis: A double–blind, placebo–controlled trial. Gastroenterology, 2000, 119, 1637.
12. Pereira S.P., Howard L.M., Muiesan P. Quality of life after liver transplantation for alcoholic liver disease. Liver Transplantation, 2000, 6, 762.



