




Метаболический синдром, проявлениями которого являются абдоми¬нальное ожирение, гиперлипидемия, артериальная гипертония, ишемическая болезнь сердца, атеросклероз, сахарный диабет 2 типа и тканевая инсулинорезистентность, приводит к патологическим изменениям в печени в 30% случаев: к жировой дистрофии печени – стеатозу или к жировой дистрофии печени с воспалением – стеатогепатиту [15–18].
Формирование стеатогепатита сопровождается воспалительно–некроти¬ческими изменениями в печени.
При избыточном бактериальном росте в кишечнике нарушаются процес¬сы ферментативного гидролиза белков и синтеза витаминов, что приводит к дефициту белков, ферментов и коферментов, участвующих в синтезе липотротеидов очень низкой плотности (ЛПОНП), и как следствие – жир не выводится из гепатоцита, а накапливается в печени. Хронический эндогенный дефицит в клетках жирных кислот приводит к изме¬нению жирнокислотного состава фосфолипидов и физико–химических свойств плазматических мембран клеток. Результатом этого являются нарушение ак¬тивности Nа,К–АТФ–азы и снижение чувствительности к инсулину. Наруше¬ние толерантности к глюкозе также является результатом этого процесса. При нарушении кишечного барьера в портальную систему печени поступают бак¬терии и их токсины, что приводит к развитию воспаления и некрозу гепатоцитов [18].
При кислородной недостаточности блокируются ферменты, участвую¬щие в синтезе липопротеидов очень низкой плотности, которые являются транспортной системой выведения жира из гепатоцита. Свободные жирные кислоты (СЖК) являются субстратом перекисного окисления липидов, актива¬ция которого приводит к развитию стеатогепатита.
Новейшие достижения в области экспериментальных и клинических ис¬следований указывают на тесную связь многих патологических состояний с процессом свободнорадикального окисления (СРО), который рассматривается как универсальный механизм повреждения клетки. Поскольку образование ра¬дикалов происходит преимущественно на мембранах клетки (в частности, гепа¬тоцита), есть основания полагать, что воспалительная и некротическая актив¬ность и синдром цитолиза, выполняющих важную роль в повреждении печени, во многом обусловлены характером регуляции СРО.
Перекисное окисление липидов в печени может приводить к образова¬нию потенциально токсичных промежуточных продуктов, которые могут вы¬звать воспалительные процессы в печени. Накопление свободных жирных ки¬слот в гепатоцитах может приводить к набуханию митохондрий, повышенной склонности к их разрушению и усилению мембранной проницаемости, что, в свою очередь, может стать причиной повышения активности аминотрансфераз [15,18,23].
В эксперименте изучали роль эндотелиальной и индуцируемой форм синтазы оксида азота в повреждении печени в процессе ишемии и пришли к выводу, что синтаза оксида азота играет важную роль в защите клеток печени от повреждающего действия [24].
Целью настоящего исследования было изучение роли оксида азота в формировании воспаления при стеатогепатите с метаболическим синдромом.
Материал и методы
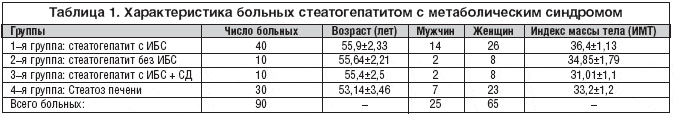
Было обследовано 90 больных стеатогепатитом с метаболическим син¬дромом, характеристика которых представлена в таблице 1. Верификацию диагноза больным проводили по данным клинических, биохимических, инструментальных методов исследования и морфологического анализа биопсийной ткани печени. Контрольную группу составили 20 здоровых доноров.
Уровень метаболитов оксида азота в сыворотке крови определяли по ме¬тоду Метельской В.А. (2005 г.). Скрининг–метод определения оксида азота достаточно точно отражает степень активности синтазы оксида азота по уров¬ню метаболитов в сыворотке крови. После депротеинизации сыворотки крови с помощью этилового спирта и центрифугирования, уровень метаболитов (суммарную концентрацию нитратов и нитритов) определяли колориметриче¬ским методом по развитию окраски в реакции диазотирования нитритом суль¬фаниламида, входящего в состав реактива Грисса. Восстановителем служил ванадия хлорид (Германия). При оценке эндогенного синтеза оксида азота прежде всего следует учитывать состав диеты. Кровь брали натощак после низконитратного ужина [7].
Актуальным представляется определение степени избыточной продук¬ции оксида азота при различных патологических состояниях, сопровождаю¬щихся воспалением (в данном случае при стеатогепатите). Концентрация окси¬да азота будет отражать активность индуцибельной изоформы NO–синтазы.
Оценка степени воспаления и фиброза с помощью УЗИ затруднена и не¬надежна [15], поэтому содержание оксида азота, как маркера воспалительной реакции, может служить дополнительным критерием оценки.
Результаты и обсуждение
Основой метаболического синдрома является нарушение структуры и функции клеточной мембраны, повреждаются сигнальная и транспортная сис¬темы, блокируется поступление в клетки глюкозы и жирных кислот. Биохими¬ческой основой метаболического синдрома является повышение в крови сво¬бодных насыщенных жирных кислот.
Индекс массы тела у больных стеатогепатитом превышал показатели нормы и был в 1–й группе в среднем 36,4 (увеличенный на 45,6%), во 2–й группе – 34,8 (увеличенный на 39,2%) и в 3–й группе – 31 (увеличенный на 24%).
Жировая ткань является источником биологически активных веществ, в том числе фактора некроза опухолей (ФНО), ключевого медиатора инсулинорезистентности, лептина–активатора окисления жирных кислот и др. [19]. При зна¬чительном увеличении массы жировой ткани развивается инсулинорезистентность. Имеются данные, что гипертрофированные адипоциты выделяют ИЛ–6, и его повышенный уровень может способствовать выработке фибриногена при висцеральном ожирении и диабете [29].
У всех больных стеатогепатитом с метаболическим синдромом выявлено достоверное повышение в плазме крови активности ферментов цитолиза и холестаза АЛТ и ACT, а также ГГТП. Показатели липидного спектра у обсле¬дуемых больных существенно изменялись: у всех было повышено общее со¬держание липидов, липопротеидов низкой плотности (ЛПНП), триглицеридов (ТГ), а уровень липопротеидов высокой плотности (ЛПВП) снижен. Уровень глюкозы был выше нормальных показателей почти в 2 раза и составлял 7,6 ммоль/л (норма 4,1–5,9 ммоль/л). Содержание холестерина повышено до 6,03 ммоль/л (норма 1,4–5,2 ммоль/л). ЛПНП: 4,8 (норма 2,3,0 ммоль/л). ЛПВП: 1,19 (норма 0,9–2,0 ммоль/л). ТГ: 2,03 (норма 0–1,7 ммоль/л). ACT: 82,9 (норма 5–35 Е/л). АЛТ: 97,5 (норма 5,5 Е/л).
Содержание стабильных метаболитов оксида азота в сыворотке крови больных стеатогепатитом с метаболическим синдромом представлено в табли¬це 2. Согласно полученным данным у больных стеатогепатитом с метабо¬лическим синдромом обнаружено достоверное повышение уровня метаболи¬тов оксида азота в 1–й группе на 42% и составило в среднем 45,83±3,57
мкмоль/л (р<0,001), во 2–й группе на 69%, 54,4±4,33 мкмоль/л, и в 3–й группе на 45,8%, в среднем 46,9±4,77 мкмоль/л (р<0,001) по отношению к контролю. У больных стеатогепатитом с сопутствующей ИБС уровень метаболитов окси¬да азота был ниже, чем в группе без ИБС, что может быть связано с эндотелиальной дисфункцией.
Усиление синтеза оксида азота может играть важную роль в защите кле¬ток печени от повреждающего действия токсических веществ. С другой сторо¬ны, избыток NO ухудшает функцию эндотелия, подавляет продукцию эндотелиального NO и угнетает сократительную функцию миокарда. Содержание метаболитов оксида азота в 3–й группе практически не отличалось от 1–й груп¬пы. По–видимому, у больных без сахарного диабета также имеются нарушения углеводного обмена, что подтверждается повышением уровня глюкозы в крови. При стеатозе печени содержание оксида азота не отличалось от контроля. Оксид азота является ключевым соединением в системе регуляции микроцир¬куляции и других жизненно важных процессов, таких как свертывание крови. Первичные медиаторы воспаления – цитокины формируют воспаление и инициируют синтез гепатоцитами комплекса вторичных медиаторов воспале¬ния [8,21]. При гипергликемии и ожирении не только гепатоциты, но и адипоциты секретируют белки острой фазы воспаления. По литературным данным, при стеатогепатите в гепатоцитах синтезируются различные медиаторы воспа¬ления, в том числе индуцибельная синтаза оксида азота; они секретируются из жи¬ровой ткани и регулируют чувствительность рецепторов к инсулину [19]. Ха¬рактерной чертой воспаления является накопление нейтральных липидов: главным образом триглицеридов в цитозоле печеночных клеток. Воспаление и дефицит жирных кислот в большинстве клеток являются причиной того, что в клетках снижается синтез холестерина. У больных атеросклерозом изменяется характер взаимодействия липопротеидов крови и биомембран эндотелия сосу¬дистой стенки, что способствует усиленному переходу холестерина в клетку. Образуются более вязкие мембраны, насыщенные холестерином.
Инфильтрация интимы кровеносных сосудов липидами происходит при каждом воспалительном процессе, независимо от этиологии. В первую очередь происходят нарушения в мембранных белках–транспортерах глюкозы, что приводит к развитию инсулинорезистентности. Дефицит в клетках жирных ки¬слот моделирует высокий потенциал воспаления, гиперкоагуляции и различ¬ных осложнений [9]. В исследованиях отмечается, что у больных с умеренной гиперхолестеринемией повышается уровень оксида азота в крови до 56±7 мкмоль/л (в контроле 35±3 мкмоль/л) [5]. По данным других исследований, у больных с атерогенным стенозом внутренней сонной артерии также имеет ме¬сто повышение уровня оксида азота до 33,0±2,9 мкмоль/л (норма 26,2±1,1 мкмоль/л) [6]. Существует тесная взаимосвязь между ожирением и дислипидемией, артериальной гипертонией, нарушенной толерантностью к глюкозе. Об¬наружена связь массы жира в организме и его расположением в абдоминальной области с маркерами хронического воспаления. Данные, полученные нами СФ методом, согласуются с данными, полученными методом ВЖХ в группах здо¬ровых доноров, больных сахарным диабетом и гипертонией, и показывают, что в образцах сыворотки крови уровень нитратов в группе здоровых доноров со¬ставляет 29,9±2,8 мкмоль/л, у больных СД – 58,6±6,9, а при АГ – 35,4±2,5 мкмоль/л [11].
У больных стеатогепатитом с метаболическим синдромом отмечается увеличение содержания метаболитов оксида азота, обусловленное повышени¬ем уровня липопротеидов низкой плотности, которые стимулируют активность индуцибельной синтазы оксида азота.
Биологические эффекты оксида азота определяются его биодоступно¬стью, а также его содержанием, утилизацией в тканях или окислением при участии СР и модифицированных ЛПНП и других соединений. Как избыток, так и дефицит оксида азота неблагоприятен для организма. Высокие концен¬трации токсичны для клеток, ферментов, вызывают модификацию белков, по¬вреждают нуклеиновые кислоты. Оксид азота и супероксидные радикалы ре¬гулируют окисление ЛПНП и приводят к их модификации [14,26]. По данным литературы, избыток оксида азота ингибирует белки–ферменты дыхательной цепи митохондрий и цикла Кребса, снижает синтез АТФ, что ведет к некрозу или апоптозу печеночных клеток.
Выводы
1. У больных с метаболическим синдромом при стеатозе печени в ре¬зультате отсутствия воспалительной реакции содержание метаболитов оксида азота не отличается от показателей контрольной группы.
2. При стеатогепатите у больных с метаболическим синдромом отмеча¬ется усиление продукции оксида азота, более выраженное у больных без ИБС.
3. Определение метаболитов оксида азота как маркера воспалительной реакции может служить дополнительным критерием оценки степени воспале¬ния и прогнозирования течения стеатогепатита.

Литература
1. Меньшикова Е.Б., Зенков Н.К., Реутов В.П. Оксид азота и NO–синтазы в организ¬ме млекопитающих при различных функциональных состояниях // Биохимия.
2000. Т. 65. Вып. 4. С. 485–503.
2. Журавлева И.А., Мелентьев И.А., Виноградов Н.А. Роль окиси азота в кардиоло¬гии и гастроэнтерологии // Клин. мед. 1997. Т. 75. № 4. С. 18–21.
3. Ковальчук Л.В., Хараева З.Ф. Роль оксида азота в иммунопатогенезе стафилокок¬ковых инфекций // Иммунология. 2003. № 3. С. 186–188.
4. Сосунов А.А. Оксид азота как межклеточный посредник // Сорос, образов, жур¬нал. 2000. Т. 6. № 12. С. 27–34.
5. Манухина Е.Б., Дауни Х.Ф., Маллет Р.Т., Малышев И.Ю. Защитные и повреж¬дающие эффекты периодической гипоксии: Роль оксида азота // Вестник Росс.
АМН. № 2. 2007. С. 25–33.
6. Голиков П.П., Леменев В.Л., Ахметов В.В. и др. Характер взаимосвязи оксида азо¬та с ангиотензинпревращающим ферментом и малоновым диальдегидом у боль¬ных с атерогенным стенозом внутренней сонной артерии // Клин, медицина. № 7. 2004. С. 15–19.
7. Метельская В.А., Туманова Н.Г. Скриннинг–метод определения уровня метаболи¬тов оксида азота в сыворотке крови // Клин, лаборат. диагн. № 6. 2005. С. 15–18.
8. Ольбинская Л.И., Игнатенко СБ. Роль системы цитокинов в патогенезе хрониче¬ской сердечной недостаточности // Тер. архив. 2001. № 1. С. 82–84.
9. Титов В.Н. Оксид азота в реакции эндотелий зависимой вазодилатации. Основы единения эндотелия и гладкомышечных клеток в паракринной регуляции метабо¬лизма. Клин, лабор. диагностика. № 2. 2007. С. 23–39.
10. Коробейникова Э.М., Кудревич Ю.В. Оценка состояния нитроксидергической вазорелаксации по содержанию нитратов в сыворотке крови больных ИБС // Клин, лабор. диагностика. № 10. 2001. С. 2–3.
11. Бондарь И.А., Климонтов В.В., Поршенников И.А. Оксид азота и диабетические ангиопатии. Сахарный диабет. 1999. № 4. С. 1–3.
12. Малышев И.Ю. Введение в биохимию оксида азота. Роль оксида азота в регуля¬ции основных систем организма // Росс, журнал гастроэнтерологии, гепатологии, колопроктологии. № 1. 1997. С. 49–55.
13. Драпкина О.М., Задорожная О.О., Ивашкин В.Т., Манухина Е.Б., Малышев И.Ю. Особенности синтеза оксида азота у больных инфарктом миокарда // Клин, меди¬цина. № 3. 2000. С. 19–23.
14. Груздева О.В. Способность липопротеинов низкой плотности к окислению и продукция кислорода и оксида азота мононуклеарными лейкоцитами больных инсулиннезависимым сахарным диабетом // Бюлл. экспер. биологии и медицины. 2001. Прил. 1.С. 21–22.
15. Подымова С.Д. Жировой гепатоз, неалкогольный стеатогепатит. Клинико–морфологические особенности. Прогноз. Лечение // МРЖ. Болезни органов пищеварения. Т. 7. № 2. 2005. С. 61–67.
16. Мельникова Н.В., Звенигородская Л.А., Хомерики С.Г., Овсянникова О.Н. Мето¬ды коррекции атерогенной дислипидемии у больных неалкогольным стеатогепатитом // МРЖ. Болезни органов пищеварения. Т. 8. № 2. 2006. С. 69–73.
17. Буеверов А.О., Богомолов П.О. Многофакторный генез жировой болезни печени // Гепатол. форум. Клинич. фармакология и терапия. 2006. № 3. С. 4–10.
18. Яковенко Э.П. Метаболические заболевания печени // Фарматека. № 10. 2003. С.31–39.
19. Корнеева О.Н., Драбкина О.М., Буеверов А.О., Ивашкин В.Т. Неалкогольная жи¬ровая болезнь печени как проявление метаболического синдрома. Клинические перспективы гастроэнтерологии, гепатологии. № 4. 2005. С. 21–24.
20. Виноградов Н.А. Многоликая окись азота // Росс, журнал гастроэнт., гепат., колопроктол. № 2. 1997. С. 6–11.
21. Царегородцева Т.М., Серова Т.И., Звенигородская Л.А., Лазебник Л.Б. Экспер. и
клин, гастроэнтер. № 1. 2007. С. 468.
22. Лазебник Л.Б., Дроздов В.Н., Барышников Е.Н. Роль NO в этиопатогенезе неко¬торых заболеваний органов пищеварения // Эксп. и клин, гастроэнт. № 2. 2005. С. 4–11.
23. Федоров И.Г., Никитин И.Г., Сторожаков Г.И. Неалкогольный стеатогепатит: клиника, патогенез, диагностика, лечение // Consilium mtdicum. 2004. V. 6. № 6. P. 401–405.
24. Kanaehi Shigeyki, Hines Jan N. // Nitric oxide synthase and postischemic liver injury // Biochem. And Biophys. Res. Commun. V. 200, 276, № 3. P. 851–854.
25. Minamiyama Yukiko. Isoforms of cytochrome P–450 on nitrate derived oxide release in Human Htart vesels // Febs. Wett. 1999. V. 452. № 3. P. 165–169.
26. Alison B. Nitric oxide regulation of free radical and enzymemediated lipid and lipoprotein oxidation // Ateriosclerosis, Thrombosis and Vase. Biol. 2000. V. 20. № 7. P. 1707–1715.
27. Moncada S. Nitric oxide and cell. Respiration Physiology and Patology. // Verk Kon. Acad. Genelsk Belg. 2000. V. 62. № 3. P. 171–179.
28. Lubrano V., Vassale С The effect of lipoproteins on endothelial nitric oxide synthase is modulated by lipoperoxides // Eur. J. Clin. Invest. 2003. V. 33. P. 117–125.
29. Festa Adostino R., Williams K., Karter A.I. The relation of body fat mass and distribu¬tion to markers of chronic inflammation // Int. J. Obesity. 2001. V. 25. № 10. P. 1407–
1415.



