




Введение. Многоформная эритема (МЭ) – это острое самоизлечивающееся заболевание кожи, характеризующееся внезапным появлением симметричных фиксированных красных узелков, часть которых превращается в типичные и/или изредка «атипичные» узелковые мишеневидные элементы. Эта сыпь часто вызывается инфекцией, особенно вирусом простого герпеса. Различают две формы МЭ – большая и малая многоформная экссудативные эритемы. Обе имеют характерным признаком одинаковый тип первичных поражений (мишени), но различаются присутствием или отсутствием поражения слизистой оболочки и общими симптомами. У подавляющего большинства больных клинически МЭ можно отличить от синдромов Стивенса–Джонсона и Лайелла, основываясь на типе первичных поражений кожи и их распределении.
История. МЭ впервые описал австрийский дерматолог Фердинанд фон Гебра в 1860 г. Заболевание, которое он описал, было легким с внезапным возникновением сотен красных узелков. Путем ежедневного наблюдения фон Гебра зарегистрировал те самые первоначальные узелки, преобразуемые в поражения с концентрическими зонами изменения цвета, которым он дал имя «мишеневидные элементы». Однако фон Гебра не описал ни продромального периода, ни поражений слизистых оболочек. Он установил, что это состояние было рецидивирующим, и упомянул «ежегодный тип», который рецидивировал каждую осень.
Позже МЭ подразделил на большую и малую формы Бернард Томас в 1950 г. Томас считал, что болезнь фон Гебры является малой формой МЭ, а тяжелый некроз слизистых оболочек с кожными высыпаниями, подобными МЭ, – большой формой заболевания. С 1950 г. много путаницы создалось вокруг определения МЭ, отчасти потому, что некоторые авторы, особенно в США, констатировали, что большая форма МЭ включает синдром Стивенса–Джонсона. Однако современные исследования прояснили ситуацию, обеспечив вескими доказательствами, что термин «малая форма МЭ» следует сохранить за болезнью фон Гебры, а термин «большая форма МЭ» следует использовать для МЭ, связанной с поражениями слизистых оболочек и общими симптомами, но не для описания синдрома Стивенса–Джонсона, т.к. последний и большая форма МЭ являются отдельными клиническими заболеваниями.
Эпидемиология. МЭ преимущественно наблюдают у молодых людей и весьма редко в детском возрасте. Приблизительно 20% всех случаев МЭ встречается в детстве. Имеется небольшое преобладание лиц мужского пола, но без расовых различий. Точный уровень заболеваемости МЭ неизвестен.
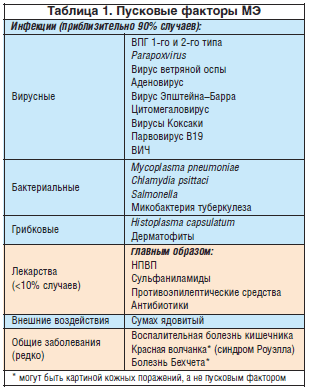
Патогенез. Современное понимание предполагает, что МЭ наиболее вероятно и у большинства больных является кожно-слизистым проявлением особой, направленной на кожу иммунной реакции, которая наблюдается в условиях инфекции у предрасположенных лиц. Вирус простого герпеса, несомненно, является самым частым связанным инфекционным фактором, а Mycoplasma pneumoniae, Histoplasma capsulatum и Parapoxvirus можно наблюдать реже. Сообщалось, что МЭ, связанная с гистоплазмозом, встречается чаще у больных с сопутствующей узловатой эритемой. В настоящее время данные за вирус Эпштейна–Барра как пусковой фактор неполны. Реже МЭ связана с лекарствами или системными заболеваниями (табл. 1). Однако необходимо учитывать возможность синдрома Стивенса–Джонсона, генерализованной фиксированной эритемы, высыпания при полиморфной лекарственной экзантеме или крапивнице, если предполагаемым диагнозом является лекарственно-индуцированная МЭ. Стоит отметить, что несколько физических факторов, например травма, холод, ультрафиолет и электронное излучение высокого напряжения, описаны как пусковые факторы МЭ, связанной с инфекциями, лекарствами или системными заболеваниями.
В настоящее время не существует четко определенной генетической предрасположенности для развития МЭ. Несколько небольших исследований связи антигенов HLA обнаружили различные ассоциации: HLA-DQw3, DRw53 и Aw33. Эти HLA-ассоциации отличаются от ассоциаций, описываемых для синдрома Стивенса–Джонсона и ТЭН.
Целый ряд местных продуктов, по сообщениям, связан с МЭ. Этот перечень включает противоинфекционные средства (хлорамфеникол, эконазол, линкомицин, неомицин, сульфаниламиды), консерванты (формальдегид, этилендиамин), сенсибилизаторы (динитрохлорбензол, дифенилциклопропанон), отдушки (перуанский бальзам), кетопрофен и местные кортикостероиды (будесонид, триамцинолона ацетонид).
Вирус простого герпеса. У большинства детей и взрослых с МЭ это заболевание вызывается ВПГ 1-го и 2-го типов. Предшествующий простой герпес губ отмечается приблизительно у 50% лиц с МЭ. Простой герпес губ может предшествовать началу поражений кожи, наблюдаться одновременно или быть явным после того, как мишеневидные элементы МЭ уже появились. Чаще всего герпес губ предшествует мишеневидным поражениям МЭ за 3–14 дней. Предполагается, что большинство случаев у детей и молодых людей обусловлено ВПГ 1-го типа, но сообщалось о подтвержденных случаях ВПГ 2-го типа у подростков и молодых людей.
Бóльшая часть наших современных знаний относительно МЭ связана с исследованием герпес-ассоциированной (ГА) МЭ. В пределах пораженного эпидермиса обнаружены не только кодированные ВПГ белки, но и ДНК ВПГ можно выявить в начальных красных узелках или внешней зоне мишеневидных элементов у 80% лиц с МЭ. Присутствие фрагментов ДНК ВПГ (чаще всего составленных из последовательностей, которые кодируют его ДНК-полимеразу) в пределах кожных поражений, а также экспрессия кодируемых вирусом антигенов на кератиноцитах может истолковываться как доказательство в пользу репликации ВПГ в пределах участков пораженной кожи. Однако репликация должна быть на низком уровне, т.к. обычно ВПГ невозможно культивировать из поражений МЭ.
Считается, что воспаление в поражениях кожи является частью ВПГ-специфичного ответа хозяина. Лица с ГАМЭ имеют нормальный иммунитет к ВПГ, но, возможно, у них есть препятствие для удаления этого вируса из инфицированных клеток; в участках пораженной кожи ДНК ВПГ может сохраняться в течение 3 мес., даже после того, как поражение уже зажило. Развитие кожных поражений запускается последовательностями ДНК ВПГ в коже и рекрутированием вирус-специфичных клеток Т-хелперов 1-го типа, которые вырабатывают γ-интерферон в ответ на вирусные антигены в коже. Считается, что за этим следует «аутоиммунный» ответ, возникающий в результате рекрутирования Т-клеток, которые отвечают на аутоантигены, высвобождаемые лизированными/апоптозными, содержащими вирусный антиген, клетками. Совсем недавно было продемонстрировано, что описанные выше фрагменты ДНК ВПГ транспортируются предшественниками клеток Лангерганса CD-34 периферической крови в места, предшествующие каждому высыпанию, где будут развиваться поражения кожи МЭ.
Клиническая картина. Из-за определенной степени клинического сходства почти до последнего времени считалось, что большая и малая форма МЭ, синдромы Стивенса–Джонсона и Лайелла являются частью спектра одного заболевания. Однако, как обсуждалось ранее, в настоящее время имеется веское доказательство в пользу того, что МЭ является заболеванием, отличным от синдромов Стивенса–Джонсона и Лайелла на нескольких уровнях – клиническом, прогностическом и этиологическом. Клинические критерии позволяют отличать обе формы МЭ от синдромов Стивенса–Джонсона и Лайелла у подавляющего большинства больных. Этими клиническими критериями являются, как следует ниже:
1. тип первичного поражения кожи;
2. распределение поражений кожи (топография);
3. присутствие или отсутствие явных поражений слизистых оболочек;
4. присутствие или отсутствие общих симптомов (табл. 2).
Первичные кожные поражения. Характерным первичным кожным поражением МЭ является типичный мишеневидный элемент. Последний имеет размер менее 3 см в диаметре, обладает правильными округлыми очертаниями и хорошо очерченным краем и состоит по крайней мере из трех отчетливых зон, например из двух концентрических колец изменения цвета, окружающих центральную круговую зону, которая имеет признаки повреждения эпидермиса в форме формирования пузыря или корки. Такой типичный мишеневидный элемент иногда называют «поражением-радужкой» из-за его радугоподобного внешнего вида.
Начальные мишеневидные элементы часто имеют центральную темную и красную наружную зоны, но могут развиваться в три зоны изменения цвета. Каждое концентрическое кольцо в пределах мишеневидного элемента, вероятнее всего, представляет собой результат того же самого продолжающегося патологического процесса. Это может объяснить, почему некоторые пациенты имеют только ограниченное количество полностью развитых типичных мишеней среди целого ряда мишеневидных элементов, которые еще не типичны или не полностью развиты, в то время как у других все эти поражения находятся на той же самой стадии развития, создавая, таким образом, мономорфное клиническое проявление. С учетом возможности того, что может присутствовать только немного типичных мишеневидных элементов, очень важен полный осмотр кожи.
При МЭ возвышающиеся атипичные узелковые мишеневидные элементы могут либо сопровождать типичные мишеневидные элементы, либо составлять первичное кожное поражение. Эти особые поражения определяются как округлые, отечные, пальпируемые и напоминающие МЭ, но только с двумя зонами и/или плохо ограниченным краем. Их необходимо отличать от плоских (пятнистых) атипичных мишеней, которые наблюдаются при синдроме Стивенса–Джонсона и ТЭН, но не при МЭ. Последние определяются как округлые поражения, которые также напоминают МЭ, но только с двумя зонами/или плохо ограниченным краем, а также являются непальпируемыми (с исключением возможного центрального пузырька или пузыря).
Распределение кожных поражений. Хотя существуют значительные колебания у разных индивидуумов, обычно присутствуют многочисленные поражения. Вообще поражения МЭ развиваются преимущественно на конечностях и лице; мишеневидные элементы, так же как и все высыпания МЭ, предпочитают верхние конечности. Тыльная поверхность кистей и предплечья является наиболее часто поражаемым местом, но ладони, шея, лицо и туловище также являются распространенными локализациями. Поражение голеней наблюдается реже. Также элементы МЭ могут появляться в пределах участков солнечных ожогов. Кроме того, высыпания имеют тенденцию группироваться, особенно на локтях и коленях. Может наблюдаться феномен Кебнера, причем мишеневидные элементы появляются на участках повреждения кожи, например при расчесах или как эритема и отек проксимальных ногтевых валиков в местах хронической самотравмы. Это повреждение должно предшествовать началу высыпания МЭ, т.к. феномен Кебнера не может проявляться, когда поражения МЭ уже появились.
Поражения слизистых оболочек. Тяжелое поражение слизистой оболочки является характерным признаком большой формы МЭ. Поражение слизистой обычно отсутствует при малой форме МЭ, а когда присутствует, поражений немного по числу и они умеренно-симптоматичны. Первичные поражения слизистой везикулобуллезны и быстро превращаются в болезненные эрозии, которые поражают слизистую губ и щек, а также слизистые глаз и половых органов. На губах эрозии быстро покрываются болезненными корками. Эрозии аногенитальной слизистой часто протяженные и полициклические с влажным основанием.
Общие симптомы. Общие симптомы почти всегда имеются при большой форме МЭ и отсутствуют или ограничены при малой. При большой форме МЭ этими общими симптомами, которые обычно предшествуют поражениям кожи или сопровождают их, являются лихорадка и слабость различных степеней. Артралгии с отеком сустава иногда описывали так же, как поражение легких, подобное атипичной пневмонии. Является ли последняя легочным проявлением МЭ, выбирая из одной из связанных инфекций, например Mycoplasma pneumoniae, неясно. Почечные, печеночные или гематологические нарушения в контексте большой формы МЭ редки.
Объединив эти четыре клинических критерия, можно сделать разграничение между большой, малой формой МЭ и синдромом Стивенса–Джонсона (табл. 2).
Естественное течение. При МЭ собирается анамнез внезапного появления кожных высыпаний, причем почти все эти поражения появляются в течение 24 ч и полного развития достигают за 72 ч. Отдельные элементы остаются фиксированными на этом же самом месте в течение 7 дней и более. Больные испытывают чувство зуда и жжения в пораженных местах.
Для большинства лиц с МЭ эпизод длится 2 нед. и заживление происходит без осложнений; одним возможным редким исключением являются глазные осложнения в условиях большой формы МЭ, которые могут наблюдаться, если немедленно не начат надлежащий уход за глазами. Иногда наблюдается поствоспалительная гипер- или гипопигментация. У больных с МЭ обычно наблюдается неосложненное клиническое течение, хотя рецидивы в случае ГАМЭ довольно распространены. Один рецидив, как было описано фон Геброй, может наблюдаться каждую осень. Большинство больных с рецидивирующей ГАМЭ имеют один или два эпизода в год, а исключением являются пациенты, получающие иммуносупрессивные препараты. Использование иммуносупрессивных средств, таких как кортикостероиды, может быть связано с более частыми и более длительными эпизодами МЭ. Эти лица могут иметь 5 или 6 эпизодов в течение года или почти непрерывное заболевание, при котором один приступ не полностью разрешился до того, как наблюдается другой. Также увеличивается частота вторичных бактериальных инфекций с длительным применением кортикостероидов.
Гистопатология. МЭ является клинико-патологическим, а не только гистологическим диагнозом. Гистологические данные характерны, но неспецифичны и используются только для исключения заболеваний в дифференциальном диагнозе, например красной волчанки и васкулита. При МЭ кератиноциты являются мишенью воспалительного повреждения, а апоптоз отдельных кератиноцитов является самой ранней патологической находкой. По мере того, как развивается процесс, наблюдаются спонгиоз и фокальная вакуольная дегенерация базальных кератиноцитов. Видны также поверхностный дермальный отек и околососудистые инфильтраты мононуклеарных лейкоцитов и Т-лимфоцитов. Иммунофлуоресцентные данные неспецифичны. Описаны гранулярные депозиты JgM и C3 вокруг поверхностных кровеносных сосудов и локально в эпидермально-дермальном соединении. Специфические антигены ВПГ выявлены в кератиноцитах иммунофлуоресценцией, геномная ДНК ВПГ установлена амплификацией ДНК-образцов биопсии кожи.
По сравнению с синдромом Стивенса–Джонсона компонент дермального воспаления более заметен при МЭ, а компонент эпидермального некролиза более изолирован. При МЭ не наблюдаются большие пласты эпидермального некроза.
Дифференциальный диагноз. Многие специалисты (недерматологи) осуществляют гипердиагностику МЭ, относя лиц с крапивницей в категорию МЭ. Необходимо строго соблюдать клинические критерии Brice и соавт. для разграничения МЭ от крапивницы (табл. 3).
Многие заболевания могут включать образование мишенеподобных элементов и мимикрировать МЭ, включая ранее упомянутую крапивницу, фиксированную лекарственную эритему, подострую кожную красную волчанку, кольцевидную центробежную эритему и несколько форм васкулитов. Чтобы исключить эти заболевания, используется биопсия кожи. Однако существует значительный клинический перекрест между МЭ и генерализованной фиксированной эритемой. Помогает в постановке диагноза выявление общего количества поражений (сотни), но важно также попытаться определить количество поражений, имевшихся при первой вспышке заболевания (т.е. меньше высыпаний при фиксированной лекарственной эритеме).
Рецидивирующая МЭ в детстве может мимикрировать в полиморфную световую реакцию или ювенильное весеннее высыпание, при которых они могут вызываться солнцем и развиваться с первым значительным воздействием солнца весной. У больных с системной красной волчанкой редкие отдельные поражения могут имитировать истинные мишеневидные поражения МЭ, но другие поражения, характерные для красной волчанки, обычно тоже присутствуют. Начальные поражения при васкулите, особенно уртикарном, могут подражать мишеневидным элементам МЭ. Требуется биопсия кожи, чтобы исключить как системную красную волчанку, так и васкулит; дополнительные ключи включают повышенную СОЭ, аутоантитела и низкие сывороточные уровни комплемента.
Лечение. Варианты терапии включают в себя общее и местное лечение острого заболевания, а также профилактические мероприятия при рецидивирующем течении. Местное лечение состоит из местных антисептиков для эрозированных кожных поражений и антисептических/антигистаминных полосканий и местных анестезирующих растворов при поражении полости рта. Назначение местных глазных средств должно осуществляться в сотрудничестве с окулистом.
Не существует двойных слепых или открытых исследований общих методов лечения для острого эпизода МЭ. Когда пусковой фактор возможно выявить (например, ВПГ или Mycoplasma pneumoniae), следует начать специфическое лечение; в случае ВПГ это будет супрессивная терапия. В большинстве случаев МЭ, особенно малой формы, обычно бывает достаточно симптоматического лечения, легкие случаи не лечат. Антигистаминные средства внутрь в течение 3–4 дней могут уменьшить жжение и зуд кожи.
Больные с множественными мишеневидными элементами и тяжелой формой МЭ с функциональными нарушениями быстро отвечают на 1–3-недельный курс преднизолона. Преднизолон (0,5–1,0 мг/кг) продолжают принимать до достижения контроля (устранения или облегчения симптомов заболевания – прим. переводчика). Вторым вариантом системного лечения глюкокортикоидами является пульс-терапия метилпреднизолоном в дозе 20 мг/кг/сут. в течение 3-х последовательных дней. Лечение с помощью преднизолона может успешно оборвать рецидив.
Ацикловир, применяемый непрерывно по 400 мг 2 раза/сут., предупреждает ГАМЭ во многих случаях. ГАМЭ не предупреждается, если ацикловир назначается после того, как рецидив простого герпеса очевиден, и не имеет никакой пользы после того, как МЭ развилась. Ацикловир использовался некоторыми пациентами непрерывно в течение многих лет без каких-либо явных побочных эффектов.
Больные рецидивирующей МЭ должны получать ацикловир (где ВПГ не является явным пусковым фактором) в течение 6 мес. Валацикловир и фамцикловир всасываются лучше, чем ацикловир, и могут использоваться у больных, которые не отвечают на ацикловир. Если эти способы лечения не имеют успеха, можно попробовать дапсон или антималярийные средства. Азатиоприн успешно использовали у пациентов с тяжелым заболеванием, у которых все другие способы лечения не дали успеха. Ответ на лечение был дозозависимым (100–150 мг/сут.). После прерывания лечения наблюдались рецидивы.
Синдром Стивенса–Джонсона
и Лайелла
Введение. Синдром Стивенса–Джонсона и синдром Лайелла – редкие, острые и угрожающие жизни заболевания кожи и слизистых оболочек, которые почти всегда являются лекарственно-обусловленными. Они являются следствием распространенной клеточной смерти кератиноцитов, которая ведет к отделению значительных участков кожи в эпидермодермальном соединении, вызывая внешний вид ошпаренной кожи. Эта распространенная клеточная гибель также ведет к отслойке слизистой оболочки, и она вносит вклад в типичные симптомы синдрома Стивенса–Джонсона и ТЭН, которые включают: высокую лихорадку, боль в коже от умеренной до сильной, страх и слабость. Заболевание имеет непредсказуемое течение. Первоначально доброкачественно выглядевший дерматоз может быстро прогрессировать, а когда видимая отслойка кожи уже произошла, то трудно определить, когда она завершится. Несколько исследований коснулись клинических признаков синдрома Стивенса–Джонсона и ТЭН, и в настоящее время существует основа диагностических критериев. Риск смерти также может быть точно спрогнозирован, для этого применяют показатель тяжести болезни, специально разработанный для прогнозирования клинического исхода ТЭН.
Т.к. прогноз соотносится с быстротой, с которой лекарство-виновник выявляется и отменяется, то крайне важно быстро установить правильный клинический диагноз с тем, чтобы причинное лекарственное средство могло быть отменено и надлежащее консервативное лечение было начато как можно быстрее. Хотя в течение длительного времени в прошлом считалось, что ТЭН и, особенно синдром Стивенса–Джонсона, являются частью спектра заболеваний, которые включали большую форму МЭ (любая из которых имеет клинически сходные поражения слизистых), эти состояния в настоящее время необходимо различать. Синдром Стивенса–Джонсона и ТЭН надо также отличать от дерматозов, таких как синдром стафилококковой обожженной кожи, генерализованная фиксированная лекарственная эритема и острый генерализованный экзентематозный пустулез, т.к. их ведение и прогноз совершенно разные.
Быстрая диагностика и, соответственно, быстрое определение и отмена причинного лекарства признаны главным фактором улучшения исхода при синдроме Стивенса–Джонсона и ТЭН. Высококачественное поддерживающее лечение, если оно возможно в блоке интенсивной терапии с современным оборудованием и обученным средним медицинским персоналом, может улучшить исход. Специфические способы лечения синдрома Стивенса–Джонсона и ТЭН еще не достигли стандартов одобрения медицины, основанной на доказательствах, отчасти потому, что истолкование молекулярных механизмов в настоящее время неполное. Их низкий уровень распространения и угрожающий жизни потенциал делают рандомизированные клинические испытания трудными к выполнению. Несмотря на это, интересные подходы, основанные на известных элементах патогенеза синдрома Стивенса–Джонсона и ТЭН и небольших сериях случаев заболевания, уже описаны.
История. В 1922 г. два американских врача, Стивенс и Джонсон, описали острый кожно-слизистый синдром у двух подростков. Это заболевание имело типичными признаками тяжелый гнойный конъюнктивит, тяжелый стоматит с обширным некрозом слизистой оболочки и подобную МЭ кожную сыпь. Оно стало известно в качестве синдрома Стивенса–Джонсона и было признано тяжелым заболеванием кожи и слизистых с редкими смертельными исходами. Синдром Стивенса–Джонсона в 1950 г. был назван Бернардом Томасом большой формой МЭ. Однако современные клинические исследования сделали очевидным, что термин «большая форма МЭ» не должен использоваться, чтобы описывать синдром Стивенса–Джонсона, т.к. это различные заболевания.
В 1956 г. Алан Лайелл описал четверых больных с некоей сыпью, «имеющей сходство с ошпариванием кожи объективно и субъективно», которую он назвал «токсический эпидермальный некролиз». «Токсический» – приписываемый токсемии – циркуляции какого-то токсина, который, как полагали, отвечает за общие симптомы и эпидермальный некролиз. Лайелл придумал термин «некролиз», скомбинировав ключевой клинический признак «эпидермолиз» с типичным гистопатологическим признаком «некроз». Он также описал тяжелое проявление болезни на слизистых оболочках как часть этого синдрома и отметил, что имелось очень слабое воспаление в дерме, признак, который позже назвали «дермальное безмолвие». Этот признак расходился с явным воспалительным инфильтратом других пузырных заболеваний, таких как МЭ, герпетиформный дерматит и буллезный пемфигоид.
Происхождение этого «токсина» не сразу стало ясным. Лекарства (сульфаниламиды и бутазоны) и виды стафилококка были основными кандидатами. В итоге такие термины, как «индуцированный стафилококком ТЭН» и «индуцированный лекарством синдром ошпаренной кожи» существовали в течение десятилетий. Когда ни лекарство, ни стафилококковая инфекция не могли быть легко определены, ТЭН назывался «смешанным» или «идиопатическим» с учетом возможных различных причин, а также отсутствия правдоподобной связи между последними и ТЭН. В итоге в то время считали, что ТЭН является типом кожной реакции на множественные стимулы.
Описание субгранулярного эпидермального расщепления у новорожденных мышей после воздействия золотистым стафилококком 2 фагогруппы, последующее открытие нового стафилококкового экзотоксина, названного эпидермолитический токсин, и отсутствие явного некроза кератиноцитов привели к разграничению ТЭН от того, который станет известен как «синдром стафилококковой ошпаренной кожи». Здесь следует отметить, что у одного из первоначальных пациентов Лайелла в действительности имелся синдром стафилококковой ошпаренной кожи, и, несмотря на то, что к тому времени он уже заметил гистологические различия между ТЭН и ССОК, эти различия были отнесены к различной степени повреждения.
По мере того, как все больше больных описывали в годы после исследования Лайелла, стало ясно, что главным образом определенные лекарства, такие как сульфаниламиды, пиразолоны, барбитураты и антиэпилептические средства, были связаны с ТЭН. В то же самое время лекарства стали рассматривать в качестве причины МЭ, связанной с тяжелым стоматитом. Поэтому в то время считалось, что эпидермальный тип МЭ (как дал определение Орфанос и соавт.), синдром Стивенса–Джонсона и ТЭН являются частью непрерывного спектра кожных реакций. Однако стало ясно, что ВПГ является главной причиной МЭ и что этот вирус не имеет отношения к ТЭН. В последнее время группа Жан-Клода Роже прояснила этот вопрос, предоставив клиническое подтверждение того, что МЭ и синдром Стивенса–Джонсона являются клинически особыми заболеваниями с разными причинами и прогнозом. Все больше появляется доказательств, что синдром Стивенса–Джонсона и ТЭН являются двумя концами диапазона тяжелых эпидермолитических побочных кожных лекарственных реакций, отличающихся только степенью отслойки кожи.
Эпидемиология. Синдром Стивенса–Джонсона и ТЭН являются редкими заболеваниями с годовым коэффициентом заболеваемости 1,2–6 и 0,4–1,2 на 1 млн населения соответственно. ТЭН поражает женщин чаще, чем мужчин, с соотношением 1,5:1, коэффициент заболеваемости увеличивается с возрастом. Группами больных с высокой степенью риска являются больные с генотипами медленных ацетиляторов, иммунокомпрометированные пациенты (например, ВИЧ-инфекция, лимфома) и больные с опухолями мозга, которые в настоящее время подвергаются радиотерапии и одновременно получают противоэпилептические средства. У лиц со СПИДом риск развития ТЭН в 1000 раз выше, чем в общей популяции.
О применении лекарств сообщается более чем у 95% больных с ТЭН. Устойчивая связь между приемом лекарства и развитием кожного высыпания наблюдается в 80% случаев. Другие редкие причины включают инфекции и иммунизацию. Литература отражает менее четкое взаимоотношение между лекарствами и синдромом Стивенса–Джонсона; утверждают, что только 50% случаев синдрома Стивенса–Джонсона являются лекарственно-обусловленными. Однако это, безусловно, является недооценкой и, наиболее вероятно, обусловлено отчасти путаницей, которая ранее существовала относительно диагностического разграничения между синдромом Стивенса–Джонсона и МЭ.
На сегодняшний день установлено, что более чем 100 лекарственных средств связаны с синдромом Стивенса–Джонсона/ТЭН. Наиболее часто участвующие лекарства перечисляются в таблице 4 и главным образом включают в себя антибиотики, нестероидные противовоспалительные препараты и антиконвульсанты. Среди первых сульфаниламиды наиболее отчетливо связаны с синдромом Стивенса–Джонсона и ТЭН; другие антимикробные средства включают аминопенициллины, хинолоны, цефалоспорины, тетрациклины и противогрибковые средства. В общем, сообщается, что риск развития синдрома Стивенса–Джонсона самый высокий в течение начальной недели (начальных недель) лечения. К тому же, более вероятно, что лекарства с длительным периодом полураспада вызовут лекарственные реакции и смертельный исход, чем средства с коротким периодами полураспада, даже если они химически родственны.
Патогенез. На сегодняшний день точная последовательность молекулярных и клеточных событий, которые ведут к развитию синдрома Стивенса–Джонсона и ТЭН, понятна только отчасти. Предполагаемый патогенез должен принять во внимание редкость этой реакции и участие специфических типов лекарств.
Убедительные факты наводят на мысль, что синдром Стивенса–Джонсона и ТЭН связаны с нарушенной способностью обезвреживать реактивные промежуточные лекарственные метаболиты. Полагают, что они инициируются иммунным ответом на антигенный комплекс, сформированный реакцией таких метаболитов с определенными тканями хозяина. Также, возможно, играет роль генетическая восприимчивость, что подтверждается повышенной частотой встречаемости HLA-B12 у лиц, которые заболели ТЭН из-за аллопуринола. Генетическая предрасположенность к синдрому Стивенса–Джонсона и ТЭН, по данным последних исследований в китайской популяции, определяется аллелем HLA-B*5801. Кроме того, сильная связь между HLA-B*5802 и индуцированным карбамазепином синдромом Стивенса–Джонсона была описана в той же самой популяции. Наконец, аллель HLA-DQB1*0601 описали у внушительно несоразмерного числа белых больных с синдромом Стивенса–Джонсона и глазными осложнениями, наводя на мысль, что эта аллель может давать повышенный риск для данного особого клинического фенотипа.
Цитотоксические Т-клетки, экспрессирующие рецептор кожного хоуминга, кожный лимфоцит-ассоциированный антиген (CTL), наблюдаются в самом начале развития кожных поражений. Вероятно, что они являются лекарственно-специфичными Т-клетками. Важные цитокины, такие как интерлейкин (ИЛ) 6, фактор некроза опухоли α, интерферон γ, ИЛ-18 и Fas лиганд, также присутствуют в пораженном эпидермисе и/или пузырной жидкости больных с ТЭН, так же как часто наблюдали несоответствие между степенью эпидермального повреждения и малочисленностью воспалительного инфильтрата. Наконец, типичный интервал между началом лекарственной терапии и синдромом Стивенса–Джонсона/ТЭН находится между 1 и 3 нед., предполагая период сенсибилизации и обеспечивая дополнительную поддержку для роли иммунной системы в их патогенезе. Этот период значительно укорачивается у больных, которые, к сожалению, подвергаются повторному воздействию лекарства, которое ранее привело к синдрому Стивенса–Джонсона или ТЭН.
В последнее время было ясно продемонстрировано, что тканевое повреждение, описанное патологами как эпидермальный некроз, обусловлено клеточной гибелью кератиноцитов посредством апоптоза. Клеточная гибель апоптозом является строго регулируемым физиологическим процессом, который делает возможным удаление ненужных клеток, не вызывая воспалительного ответа и его последствий. Нарушения в контроле апоптоза обнаруживаются при целом ряде болезней человека, например, раке, аутоиммунных нарушениях, дегенеративных заболеваниях и СПИДе.
Апоптоз кератиноцитов – явно признак ранних стадий синдрома Стивенса–Джонсона и ТЭН, он является первым характерным морфологическим признаком специфического тканевого повреждения при этом заболевании. Классическая гистологическая картина распространенного эпидермального некролиза является в действительности картиной последствия апоптоза кератиноцитов. В самом деле, это апоптотическое состояние клеток является кратковременным по природе. В физиологических условиях апоптотические клетки быстро удаляются на ранней стадии фагоцитами, причем последние имеют способность специфически выявлять и поглощать апоптотические клетки.
В ситуации, когда уровень распространенности апоптоза превышает функциональную возможность фагоцитов удалять такие клетки, эти апоптотические клетки прогрессивно некротизируются и высвобождают внутриклеточное содержимое, запуская, таким образом, воспалительный ответ. При синдроме Стивенса–Джонсона и ТЭН в течение нескольких часов апоптоз кератиноцитов становится очень обильным в пораженной коже, таким образом быстро превышая фагоцитарную функциональную способность профессиональных и непрофессиональных (например кератиноцитов), локализованных в коже. В течение промежутка от нескольких часов до нескольких дней эти апоптотические кератиноциты некротизируются; наряду с потерей сцепления к соседним кератиноцитам и базальной мембране весь эпидермис теряет жизнеспособность, создавая, таким образом, знакомую картину полнослойного эпидермального некролиза.
Определенные цитокины семейства ФНО, связавшись с их специфическими рецепторами клеточной поверхности («рецепторы смерти»), имеют способность вызывать апоптоз. Такие «рецепторы смерти» действуют как сенсоры клеточной поверхности, которые выявляют наличие особых внеклеточных сигналов смерти и быстро запускают клеточную деструкцию через апоптоз. Одним из этих клеточных сенсоров и триггеров апоптоза является так называемая пара рецептор-лиганд Fas(CD95) – Fas ligand (FasL, CD95L). Кожа является первым местом тканевого повреждения в начале синдрома Стивенса–Джонсона и ТЭН, и известно, что как Fas, так и FasL, экспрессируются в эпидермальных кератиноцитах. Недавно было продемонстрировано, что апоптоз кератиноцитов в пораженной коже больных ТЭН связан с сильно увеличенной экспрессией кератиноцитарного FasL наряду с сохраненными уровнями экспрессии кератиноцитарного Fas. Приготовление криостатных срезов пораженной кожи с Fas-чувствительными клетками в качестве мишеней продемонстрировало, что кератиноцитарный FasL цитолитически активен при ТЭН; цитолиз можно предупредить моноклональными антителами, которые препятствуют взаимодействию Fas и FasL.
Новая модель состоит в том, что в нормальной коже низкие уровни FasL экспрессируются кератиноцитами и локализуются внутриклеточно. В пораженной ТЭН коже высокие уровни FasL экспрессируются кератиноцитами и локализуются на клеточной поверхности. В итоге, взаимодействия клеточной поверхности между кератиноцитарными Fas и FasL на соседних клетках затем возможны. После контакта с Fas, FasL клеточной поверхности вызывает полимеризацию Fas и быстрое сигнализирование кератиноцитарной клеточной смерти через апоптоз. Т.к. Fas и FasL коэкспрессируются на большом количестве кератиноцитов в пораженной коже, то апоптоз кератиноцитов может быть обильным, приводя к деструкции больших участков эпидермиса.
Кроме того, некроз кератиноцитов может опосредоваться лекарственно-специфичными CD8 цитотоксическими Т-клетками через перфорин-гранзимовый путь. Ключевая роль лекарственно-специфичных Т-лимфоцитов в механизмах большинства лекарственных реакций подтверждена исследованиями in vitro многих клонов Т–лимфоцитов. CD3+, CD8+, CD28-, KIR/KAR, CLA-положительные цитотоксические Т-клетки с цитотоксичностью, подобной натуральным киллерам, преобладали в ранней пузырной жидкости ТЭН.
Индуцибельную синтазу азотной кислоты можно обнаружить в коже при синдроме Стивенса–Джонсона/ТЭН, что может указывать на то, что закись азота вносит вклад в апоптоз и некроз. Матриксная металлопротеиназа ММР2 играет значительную роль в отслойке эпидермиса, воспалении и реэпителизации.
Клиническая картина. Начальными симптомами как ТЭН, так и синдрома Стивенса–Джонсона могут быть лихорадка, жжение глаз и боль после глотания, любой из них может предшествовать кожным проявлениям за 1–3 дня. Кожные поражения имеют тенденцию сначала появляться на туловище, распространяясь на шею, лицо и проксимальные части верхних конечностей. Дистальные части рук, а также голени относительно щадятся, но ладони и подошвы могут быть начальными местами поражения. Эритема и эрозии на слизистых оболочках щек, глаз и половых органов имеются более чем у 90% больных. Эпителий дыхательного тракта поражается у 25% больных с ТЭН, также могут встречаться и желудочно-кишечные поражения (например, эзофагит, понос). Кожные высыпания обычно болезненны, особенно эрозии слизистых. Дополнительные общие проявления включают лихорадку, лимфаденопатию, гепатит и цитопении.
Морфология кожных поражений была исследована обстоятельно. Сначала поражения выглядят как эритематозные, темно-красные и пурпурозные пятна разного размера и формы и имеют склонность сливаться. На этой стадии и в присутствии поражения слизистой оболочки и болезненности должен подозреваться риск быстрого прогрессирования в синдром Стивенса–Джонсона и ТЭН. В отсутствие самопроизвольной эпидермальной отслойки симптом Никольского необходимо проверять, вызвав тангенциальное механическое давление с помощью пальца на нескольких эритематозных зонах. Этот признак считается положительным, если вызывается эпидермально-дермальное расщепление. У некоторых больных пятнистые высыпания в начале могут иметь темный центр, придавая им мишенеподобный внешний вид. Однако такие поражения не имеют трех концентрических колец, характерных для типичных мишеневидных поражений (как первоначально было описано Геброй), и не являются папулезными, как при атипичных мишеневидных поражениях МЭ.
По мере того, как поражение эпидермиса прогрессирует по направлению в сторону полнослойного некроза, эти пятнистые темно-красные высыпания принимают характерный серый оттенок. Данный процесс может быть очень быстрым (часы) или занимать несколько дней. Некротический эпидермис затем отделяется от подлежащей дермы, вызывая появление пузырей. Эти пузыри имеют особые признаки: они легко разрываются (дряблые) и могут распространяться в сторону легким давлением большого пальца, т.к. некротический эпидермис сдвигается латерально (симптом Асбо-Гансена). Кожа имеет сходство с влажной папиросной бумагой, к тому же отрывается травмой, обнажая большие участки чувствительной и кровоточащей дермы, которую называют ошпаренной. По этой причине к данным больным необходимо прикасаться с предельной осторожностью. Напряженные пузыри обычно наблюдаются только на ладонно-подошвенных поверхностях, где эпидермис толще и по этой причине устойчивее к небольшой травме.
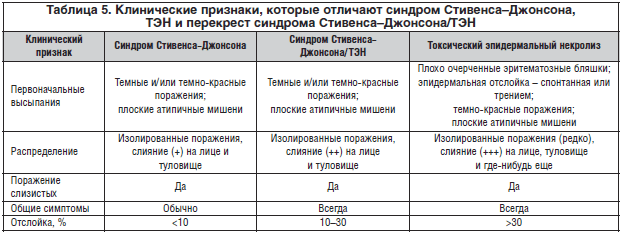
Принимая таких больных, необходимо тщательно и правильно оценивать протяженность некролиза, т.к. он является главным прогностическим фактором. Правила, обычно используемые для оценки площади поверхности термических ожогов, подходят и для этой цели. Опыт показал, что протяженность отслойки кожи легко переоценивается. Измерение должно включать отделенный и отделяемый эпидермис (положительный симптом Никольского), но не только на эритематозных участках (отрицательный симптом Никольского). Определение протяженности отслойки кожи дает возможность отнесения больных в одну из трех групп (табл. 5):
– синдром Стивенса–Джонсона: менее 10% площади поверхности тела;
– перекрест синдрома Стивенса–Джонсона – ТЭН: 10–30% площади поверхности тела;
– ТЭН: больше 30% площади поверхности тела.
Эрозии слизистых оболочек присутствуют более чем у 90% больных. Пациенты жалуются на светобоязнь и болезненное мочеиспускание.
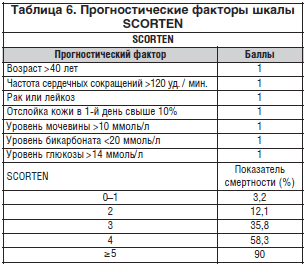
При ТЭН имеется несколько факторов, которые находились в связи с неблагоприятным исходом, включая увеличение возраста и протяженность эпидермальной отслойки. Кроме того, количество лекарств, подъем уровней сывороточной мочевины, креатинина и глюкозы, нейтропения, лимфоцитопения и тромбоцитопения были статистически связаны с неблагоприятным исходом. Поздняя отмена причинного лекарства также связана с менее благоприятным исходом. Приблизительно оценили, что быстрая отмена лекарства-аллергена уменьшает риск смерти на 30% в сутки. В последнее время был предложен подсчет баллов тяжести болезни для ТЭН (SCORTEN), в котором семь параметров с одинаковой значимостью были объединены так, чтобы сделать возможным предсказать исход (табл. 6).
В среднем, смерть наблюдается у каждого третьего больного с ТЭН и главным образом обусловлена инфекциями (золотистый стафилококк и синегнойная палочка). Массивная трансэпидермальная потеря жидкости связана с электролитным дисбалансом, угнетением секреции инсулина, инсулинорезистентностью, начало гиперкатаболического состояния также может быть способствующим фактором. Все эти осложнения ТЭН (которые также можно наблюдать при синдроме Стивенса–Джонсона) лучше лечатся в блоке интенсивной терапии. К сожалению, осложнения могут завершиться у взрослых респираторным дистресс-синдромом и полиорганной недостаточностью, несмотря на адекватное поддерживающее лечение.
Заживление участков отслоенного эпидермиса посредством реэпителизации обычно начинается через несколько дней и завершается в большинстве случаев в течение 3 нед. Этот процесс происходит в результате пролиферации и миграции кератиноцитов из резервных мест, таких как здоровый эпидермис, окружающий обнаженные участки и волосяные фолликулы в пределах участков отслойки. Благодаря этой сохраненной способности для реэпителизации при синдроме Стивенса–Джонсона и ТЭН пересадка кожи не требуется. Однако, к сожалению, заживление может быть с изъяном, и выжившие могут иметь последствия, такие как симблефарон, конъюнктивальные синехии, эктропион, вросшие ресницы, рубцевание кожи, неравномерная пигментация, эруптивные меланоцитарные невусы, длительно существующие эрозии слизистых оболочек, фимоз, вагинальные синехии, дистрофия ногтей и диффузная алопеция. Часто эти последствия можно сделать минимальными наилучшим уходом за кожей, но при ТЭН до 35% выживших могут иметь глазные симптомы, колеблющиеся от синдрома сухих глаз до слепоты.
Распознавание лекарства-аллергена является важной и трудной задачей, но оно должно быть среди первых приоритетов. Как было отмечено ранее, запоздалая отмена причинного лекарства связана с увеличенной смертностью. В настоящее время не существует надежного in vitro теста для быстрого распознавания предполагаемых лекарств. Накожные аллергические пробы демонстрируют слабую чувствительность при синдроме Стивенса–Джонсона/ТЭН и не уместны для целей распознавания; повторное воздействие на больного предполагаемым причинным лекарством является, очевидно, недопустимым способом при таких тяжелых реакциях. По этой причине клиницист вынужден полагаться на ранее сообщенные связи и устанавливать вероятность (маловероятно, вероятно, вполне убедительно, правдоподобно и т.д.) для каждого лекарства, основанную на присущей ему способности вызывать синдром Стивенса–Джонсона/ТЭН и внешних факторах, например, начала какого-то даваемого средства относительно начала синдрома Стивенса–Джонсона/ТЭН. Синдром Стивенса–Джонсона и ТЭН обычно наблюдаются через 7–21 день после начала применения причинного лекарства в условиях первого воздействия лекарством, но может наблюдаться в течение 2 дней в случае повторного воздействия лекарством, которое ранее послужило причиной синдрома Стивенса–Джонсона или ТЭН. В общем, перечень лекарств больного с синдромом Стивенса–Джонсона и ТЭН должен сокращаться к абсолютному минимуму, необходимо производить подходящие замены и предпочитать лекарства с коротким периодом полураспада.
Гистопатология. Гистопатологическое исследование пораженной кожи является весьма полезным способом для подтверждения диагноза синдрома Стивенса–Джонсона и ТЭН, т.к. морфологические данные отличаются от данных, наблюдаемых при стафилококковом синдроме ошпаренной кожи (подроговой пузырь с расслоением, локализованным в зернистом слое эпидермиса) и остром генерализованном экзантематозном пустулезе (обильный нейтрофильный инфильтрат, поверхностные эпидермальные пустулы и спонгиоз, но не полнослойный эпидермальный некролиз). Часто экстренный анализ замороженных криостатных срезов достаточен для этой цели.
В начальных поражениях при синдроме Стивенса–Джонсона и ТЭН апоптотические кератиноциты наблюдаются рассеянными в базальном и ближайших супрабазальных слоях эпидермиса. Весьма вероятно, что это гистологическое соответствие темному и серому цвету, который клиницисты, знакомые с синдромом Стивенса–Джонсона/ТЭН, рассматривают в качестве предупреждающего симптома надвигающегося полномасштабного эпидермального некролиза и отслойки. На более поздних стадиях биопсийные образцы пораженной кожи демонстрируют субэпидермальный пузырь с вышележащим сливающимся некрозом всего эпидермиса и редкий периваскулярный инфильтрат, составляемый главным образом лимфоцитами. На иммунопатологическом уровне изменчивые количества лимфоцитов (обычно CD8+) и макрофагов наблюдаются в эпидермисе, в то время как лимфоциты в сосочковом слое дермы являются в основном CD4+-клетками.
Дифференциальный диагноз. Дифференциальный диагноз синдрома Стивенса–Джонсона и ТЭН главным образом состоит из МЭ, синдрома стафилококковой ошпаренной кожи, острого генерализованного экзантематозного пустулеза и фиксированной лекарственной эритемы. Паранеопластическая пузырчатка, лекарственно-индуцированный линейный JgA буллезный дерматоз, болезнь Кавасаки, красная волчанка и тяжелая острая реакция «трансплантат против хозяина» могут также рассматриваться у некоторых больных в зависимости от клинических условий.
Гистопатологическая картина таких состояний, как термические ожоги, фототоксичность и пузыри коматозного состояния включает в себя полнослойный эпидермальный некроз, но клинико-патологической корреляции нет. В особенности распределение высыпаний и анамнез больного несовместимы с ТЭН.
Лечение. Наилучшее медицинское ведение синдрома Стивенса–Джонсона и ТЭН требует ранней диагностики, немедленной отмены причинного лекарства, поддерживающего и специфического лечения (табл. 7).
Поддерживающее лечение подобно таковому, применяемому при тяжелых термических ожогах, и нацелено на уменьшение связанных осложнений, которые являются главной причиной смертности. Осложнения включают гиповолемию, электролитный дисбаланс, почечную недостаточность и сепсис. Осторожная ежедневная обработка ран, гидратация и парентеральное питание необходимы и, предпочтительно, выполняются в блоке интенсивной терапии, если имеется отслойка эпидермиса, поражающая 10–20% (или более) площади поверхности тела. Рекомендуется использование вместо обычной кушетки и простыней терморегулируемых с управляемым давлением кушеток и простыней выживания из тонколистового алюминия. Все манипуляции с больными должны выполняться в стерильных условиях, а венозные катетеры необходимо устанавливать, если возможно, в места непораженной кожи.
Обработку ран лучше выполнять один раз в день с помощью или в присутствии дерматолога. Манипуляций на теле пациента должно выполняться как можно меньше, т.к. каждое движение является возможной причиной отслойки кожи. Уход за кожей должен сосредоточиться на лице, на области глаз, носа, рта, ушей, в аногенитальной области, подмышечных складках и межпальцевых промежутках. Неотслоенные участки кожи сохраняют сухими и над ними не манипулируют. Отслоенные участки, особенно на спине и в местах давления в соприкосновении с кушеткой, необходимо покрывать марлей с вазелином, до тех пор, пока не произошла реэпителизация. На лице серозные и/или кровянистые корки можно удалять с помощью стерильного изотонического раствора натрия хлорида. Антибактериальные мази (например, мупироцин) следует накладывать вокруг естественных отверстий (ушей, носа, рта), а силиконовые повязки можно применять, чтобы покрывать эрозированные обнаженные участки кожи. Силиконовые повязки не требуют замены и могут оставаться на месте до тех пор, пока не происходит реэпителизация, но ее поверхность необходимо ежедневно очищать стерильным изотоническим раствором хлорида натрия. Еще один способ состоит в том, чтобы поместить большую нелипкую слоистую повязку (многослойные раневые покрытия из полиэтилена, вискозы, искусственного шелка, хлопка) на больного и на кушетку.
Рекомендуется квалифицированный осмотр офтальмолога. Веки необходимо ежедневно мягко очищать с помощью стерильного изотонического раствора натрия хлорида, а глазные мази с антибиотиками накладывать на веки. Кроме того, глазные капли с антибиотиком должны применяться 3 раза/сут. на роговицу, чтобы уменьшить бактериальную колонизацию, которая может вести к рубцеванию. Ноздри нужно очищать ежедневно стерильным ватным валиком, увлажненным стерильным изотоническим раствором натрия хлорида, и далее ту же самую процедуру применить повторно, чтобы нанести немного мази с антибиотиком. Рот необходимо прополаскивать несколько раз в день, используя шприц со стерильным изотоническим раствором натрия хлорида, а затем отсасывать, если больной без сознания. В аногенитальной области и межпальцевых промежутках уход за кожей выполняется ежедневно кратковременным прикладыванием 0,5% раствора нитрата серебра в случае мацерации или просто стерильного раствора натрия хлорида, если мацерация отсутствует.
На сегодняшний день никакие специфические способы лечения синдрома Стивенса–Джонсона не достигли стандартов одобрения медицины, основанной на доказательствах. В общем, лечение для тяжелопораженных больных с синдромом Стивенса–Джонсона отобразило лечение для ТЭН, тогда как больные с более легкими формами синдрома Стивенса–Джонсона могут получать только поддерживающее лечение. Низкий уровень распространения синдрома Стивенса–Джонсона и ТЭН делает рандомизированные клинические испытания трудными к выполнению. Как следствие, литература по большей части состоит из историй болезни и небольших неконтролируемых случаев. В таких исследованиях несколько способов лечения, включая циклоспорин (3–4 мг•кг/сут.), циклофосфамид (100–300 мг/сут.), плазмаферез и N-ацетилцистеин (2,0 г/6 ч), уже продемонстрировали многообещающие результаты.
По-прежнему существует значительная полемика относительно использования системных кортикостероидов для спектра синдрома Стивенса–Джонсона и ТЭН. Современные тенденции по отношению к применению циклоспорина и внутривенного иммуноглобулина для этих больных уже сняли некоторую остроту данной дискуссии. Ряд исследований поддерживает стандартную глюкокортикостероидную терапию, однако большинство исследований представляют данные за стандартное использование ожогового центра в отсутствие глюкокортикостероидной терапии. Эти исследования сообщают о более высоком уровне смертности, особенно от сепсиса, у больных, леченных кортикостероидами, по сравнению с больными, которых вели в ожоговых центрах в отсутствие лечения глюкокортикоидами. В исследовании одного ожогового центра применение системных кортикостероидов не было связано с более высокой заболеваемостью и смертностью.
Сторонники стандартного применения системных кортикостероидов предполагают, что для больных синдромом Стивенса–Джонсона и ТЭН применение данных препаратов в начальной стадии и в течение болезни (до значительной отслойки кожи) с последующим быстрым снижением может быть успешным и даже жизнеспасающим. После того как наблюдается распространенная отслойка, риск инфекции, несомненно, перевешивает возможную пользу кортикостероидов. Важно, чтобы лекарственные и инфекционные провокаторы были установлены и удалены, если возможно. Если предписывается лечение глюкокортикоидами, то дозы до 2–2,5 мг/кг ежедневно внутривенного метилпреднизолона в разделенной дозировке первоначально обычно применяются с относительно быстрым снижением к средним дозам, когда прекращается образование новых пузырей. Еще одним способом лечения кортикостероидами может быть пульс-терапия дексаметазоном в дозе 1,5 мг/кг в течение трех последовательных дней.
В конце 1990-х гг. было два в значительной степени сбивающих с толку, если не парадоксальных, исследования по этому вопросу. По данным одного исследования, глюкокортикоидное лечение по другим показаниям, предшествовавшее началу синдрома Стивенса–Джонсона или ТЭН, не уменьшило риск этих заболеваний. Другое исследование отметило увеличенный риск синдрома Стивенса–Джонсона или ТЭН у больных, предварительно принимавших системные глюкокортикоиды.
В теории методы лечения, которые обладают способностью избирательно блокировать апоптоз кератиноцитов, имеют значительный потенциал для лечения синдрома Стивенса–Джонсона и ТЭН. Внутривенный иммуноглобулин содержит антитела против Fas-рецептора, который может блокировать молекулярное взаимодействие Fas-Fas ligand и, следовательно, апоптоз кератиноцитов.
В настоящее время многочисленные истории болезни и семь дополнительных неконтролируемых клинических исследований (которые включали по крайней мере 9 больных) проанализировали лечебный эффект внутривенного иммуноглобулина при ТЭН. Взятые вместе, несмотря на то, что каждое исследование имеет свои потенциальные систематические ошибки, пять из семи таких исследований указали на пользу (т. е. уменьшение смертности), когда внутривенный иммуноглобулин использовали в общей дозе >2 г/кг в течение 3–4 дней.
В настоящее время рекомендуемым режимом лечения является 1 г/кг/сут. внутривенного иммуноглобулина в течение трех последовательных дней, давая, таким образом, общую дозу 3 г/кг. Высокие дозы внутривенного иммуноглобулина, по-видимому, являются целесообразным и безопасным выбором среди специфических способов лечения, имеющихся в настоящее время и предварительно прошедших испытание. Однако требуются дальнейшие испытания, чтобы установить его эффективность.




Литература
1. Rook's Textbook of dermatology, eighth edition, edited by Tony Burns, Stephen Breathnach, Neil Cox and Christopher Griffiths in four volumes. Willey-Blackwell, 2010.
2. Comprehensive dermatologic drug therapy, second edition. Stephen E. Wolverton. Saunders, 2007.
3. Clinical dermatology, fifth edition. Thomas P. Habif. Mosby, 2010.
4. Dermatology, third edition, 2-volume set, edited by Jean L Bolognia MD, Joseph L Jorizzo MD, Julie V Schaffer, . Elsevier, 2012
5. Pediatric dermatology, fourth edition, 2-volume set, edited by Lawrence A. Schachner, Ronald C. Hansen. Mosby, 2011.



