



.gif)
–§–µ–љ–Њ–Љ–µ–љ–Њ–ї–Њ–≥–Є—П –і–Є—Б–њ–ї–∞–Ј–Є–є
—Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є
–°–Њ—Б—В–∞–≤–ї—П—П –Њ–Ї–Њ–ї–Њ 50% –Љ–∞—Б—Б—Л —В–µ–ї–∞, —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–∞—П —В–Ї–∞–љ—М (–°–Ґ) —П–≤–ї—П–µ—В—Б—П –Њ–і–љ–Є–Љ –Є–Ј —З–µ—В—Л—А–µ—Е –Њ—Б–љ–Њ–≤–љ—Л—Е —В–Є–њ–Њ–≤ —В–Ї–∞–љ–Є –≤ —В—А–∞–і–Є—Ж–Є–Њ–љ–љ—Л—Е –Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є—П—Е (–≤ –і–Њ–њ–Њ–ї–љ–µ–љ–Є–µ –Ї —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ–Њ–є, –Љ—Л—И–µ—З–љ–Њ–є –Є –љ–µ—А–≤–љ–Њ–є —В–Ї–∞–љ–Є). –Ю—Б–љ–Њ–≤–љ–∞—П —Д—Г–љ–Ї—Ж–Є—П –°–Ґ – —Н—В–Њ —Б—В—А—Г–Ї—В—Г—А–љ–∞—П –њ–Њ–і–і–µ—А–ґ–Ї–∞ –і—А—Г–≥–Є—Е —В–Ї–∞–љ–µ–є. –•—А—П—Й –Є –Ї–Њ—Б—В—М —П–≤–ї—П—О—В—Б—П –Њ—Б–љ–Њ–≤–љ—Л–Љ–Є —А–∞–Ј–љ–Њ–≤–Є–і–љ–Њ—Б—В—П–Љ–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є, –і—А—Г–≥–Є–µ —В–Є–њ—Л –≤–Ї–ї—О—З–∞—О—В –∞—А–µ–Њ–ї—П—А–љ—Г—О —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ—Г—О —В–Ї–∞–љ—М, —Б–Ї—А–µ–њ–ї—П—О—Й—Г—О –Њ—А–≥–∞–љ—Л, –Є –њ–ї–Њ—В–љ—Г—О —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ—Г—О —В–Ї–∞–љ–Є, —Д–Њ—А–Љ–Є—А—Г—О—Й—Г—О —Б–≤—П–Ј–Ї–Є –Є —Б—Г—Е–Њ–ґ–Є–ї–Є—П.
–Э–µ–і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–∞–љ–љ–∞—П –і–Є—Б–њ–ї–∞–Ј–Є—П —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є (–љ–Ф–°–Ґ) –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є —А–∞–Ј–љ–Њ—А–Њ–і–љ—Г—О –≥—А—Г–њ–њ—Г –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –Ї–Њ—В–Њ—А—Л–µ, –≤ —Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –Љ–Њ–≥—Г—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї —А–∞–Ј–ї–Є—З–љ—Л–Љ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ –±–Њ–ї–µ–Ј–љ—П–Љ. –љ–Ф–°–Ґ —З–∞—Б—В–Њ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В –∞–±–љ–Њ—А–Љ–∞–ї—М–љ—Л–Љ —Б—В—А—Г–Ї—В—Г—А–љ—Л–Љ –Є —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ CT. –≠—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї –љ–∞—А—Г—И–µ–љ–Є—П–Љ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є–Є –Є —Д—Г–љ–Ї—Ж–Є–є –Њ—А–≥–∞–љ–Њ–≤ [1–3]. –Ъ–ї–Є–љ–Є–Ї–Њ––Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –љ–Ф–°–Ґ –љ–µ–Њ–±—Л—З–∞–є–љ–Њ —А–∞–Ј–љ–Њ–Њ–±—А–∞–Ј–љ—Л. –Ю–љ–Є –Љ–Њ–≥—Г—В –≤–Ї–ї—О—З–∞—В—М —Б–Ї–µ–ї–µ—В–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б –љ–∞—А—Г—И–µ–љ–Є–µ–Љ —Б—В—А–Њ–µ–љ–Є—П —Е—А—П—Й–∞, –љ–µ–њ—А–Њ–њ–Њ—А—Ж–Є–Њ–љ–∞–ї—М–љ–Њ –і–ї–Є–љ–љ—Л–µ –Ї–Њ–љ–µ—З–љ–Њ—Б—В–Є, –∞—А–∞—Е–љ–Њ–і–∞–Ї—В–Є–ї–Є—О, –і–µ—Д–Њ—А–Љ–∞—Ж–Є–Є –≥—А—Г–і–љ–Њ–є –Ї–ї–µ—В–Ї–Є, —Б–Ї–Њ–ї–Є–Њ–Ј—Л –њ–Њ–Ј–≤–Њ–љ–Њ—З–љ–Є–Ї–∞, –њ–ї–Њ—Б–Ї–Њ—Б—В–Њ–њ–Є–µ, –њ–∞—В–Њ–ї–Њ–≥–Є—О —А–∞–Ј–≤–Є—В–Є—П –Ј—Г–±–Њ–≤, –њ—А–Є–Ї—Г—Б–∞, –Ї–Є—Б—В—Л, –њ–∞—В–Њ–ї–Њ–≥–Є—О —Б—Г—Б—В–∞–≤–Њ–≤ (—Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М –Ї –≤—Л–≤–Є—Е–∞–Љ), –≥–Є–њ–µ—А—Н–ї–∞—Б—В–Є—З–љ–Њ—Б—В—М, –Є—Б—В–Њ–љ—З–µ–љ–Є–µ, —Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М –Ї —В—А–∞–≤–Љ–∞—В–Є–Ј–∞—Ж–Є–Є –Ї–Њ–ґ–Є, —А–∞—Б—И–Є—А–µ–љ–Є–µ –≤–µ–љ –Є –≤–љ–µ—И–љ–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є —Г—Б–Ї–Њ—А–µ–љ–љ–Њ–≥–Њ —Б—В–∞—А–µ–љ–Є—П – —А–∞–љ–љ–µ–µ —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ –Љ–Њ—А—Й–Є–љ, –і–µ—Д–Њ—А–Љ–∞—Ж–Є—П –Њ–≤–∞–ї–∞ –ї–Є—Ж–∞, –≤ —В–Њ–Љ —З–Є—Б–ї–µ —В.–љ. –≥—А–∞–≤–Є—В–∞—Ж–Є–Њ–љ–љ—Л–є –њ—В–Њ–Ј (–Њ–±–≤–Є—Б–∞–љ–Є–µ –Љ—П–≥–Ї–Є—Е —В–Ї–∞–љ–µ–є –ї–Є—Ж–∞). –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Ф–°–Ґ –њ—А–µ–і—А–∞—Б–њ–Њ–ї–∞–≥–∞–µ—В –Ї –±—А–Њ–љ—Е–Њ–ї–µ–≥–Њ—З–љ—Л–Љ –Є —А–µ–љ–Њ–≤–∞—Б–Ї—Г–ї—П—А–љ—Л–Љ –њ–∞—В–Њ–ї–Њ–≥–Є—П–Љ, —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –њ–Њ—В–µ—А–µ –Љ—Л—И–µ—З–љ–Њ–є –Љ–∞—Б—Б—Л (–≤ —В–Њ–Љ —З–Є—Б–ї–µ —Б–µ—А–і–µ—З–љ–Њ–є –Є –≥–ї–∞–Ј–Њ–і–≤–Є–≥–∞—В–µ–ї—М–љ–Њ–є –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А—Л, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї –Ї–∞—А–і–Є–Њ–≤–∞—Б–Ї—Г–ї—П—А–љ—Л–Љ –Є –Њ—Д—В–∞–ї—М–Љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –њ–∞—В–Њ–ї–Њ–≥–Є—П–Љ [4]) –Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ —Д—Г–љ–Ї—Ж–Є–Є –ґ–µ–ї—Г–і–Њ—З–љ–Њ––Ї–Є—И–µ—З–љ—Л—Е –Њ—А–≥–∞–љ–Њ–≤ [5–7]. –Я–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–Є—Б—В–µ–Љ—Л –≤–µ—Б—М–Љ–∞ —А–∞–Ј–љ–Њ–Њ–±—А–∞–Ј–љ—Л: –њ—А–Њ–ї–∞–њ—Б –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ (–љ–∞–Є–±–Њ–ї–µ–µ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–∞—П –Є–Ј –≤—Б–µ—Е —Б–µ—А–і–µ—З–љ—Л—Е –∞–љ–Њ–Љ–∞–ї–Є–є –њ—А–Є –Ф–°–Ґ [1] –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–µ—В—Б—П, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –њ—А–Є –≠—Е–Њ–Ъ–У –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є), –≤–µ–љ–Њ–Ј–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М, –≤–∞—А–Є–Ї–Њ–Ј–љ–∞—П –±–Њ–ї–µ–Ј–љ—М [8,9], –∞ —В–∞–Ї–ґ–µ –њ–∞—В–Њ–ї–Њ–≥–Є–Є –≥–µ–Љ–Њ—Б—В–∞–Ј–∞ [10].
–Ф–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞ –Ф–°–Ґ –±–∞–Ј–Є—А—Г–µ—В—Б—П –љ–∞ —Н—В–Є—Е —Б–Є–Љ–њ—В–Њ–Љ–∞—Е –Є –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л—Е –і–∞–љ–љ—Л—Е (–љ–∞–њ—А–Є–Љ–µ—А, –∞–љ—В—А–Њ–њ–Њ–Љ–µ—В—А–Є—П, –≤–љ–µ—И–љ–µ–µ –і—Л—Е–∞–љ–Є–µ, —Г–Љ–µ–љ—М—И–µ–љ–љ—Л–є —А–∞–Ј–Љ–µ—А —Б–µ—А–і—Ж–∞, —Б–љ–Є–ґ–µ–љ–љ–Њ–µ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–µ –і–∞–≤–ї–µ–љ–Є–µ, –њ–ї–µ—В–Є–Ј–Љ–Њ–≥—А–∞—Д–Є—П, —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Є –≠–Ъ–У –Є —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–≥–Њ —Д–ї–µ–±–Њ—Б–Ї–∞–љ–Є—А–Њ–≤–∞–љ–Є—П) [8,11]. –°–Њ–≥–ї–∞—Б–љ–Њ –∞–љ–∞–ї–Є–Ј—Г —Н—В–Є—Е —Д–µ–љ–Њ—В–Є–њ–Є—З–µ—Б–Ї–Є—Е –Љ–∞—А–Ї–µ—А–Њ–≤ –љ–Ф–°–Ґ –µ–µ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–Є–µ –Љ–Њ–ґ–µ—В –±—Л—В—М —Б—А–∞–≤–љ–Є—В–µ–ї—М–љ–Њ –±–Њ–ї—М—И–Є–Љ —Б—А–µ–і–Є –Њ–±—Й–µ–≥–Њ –љ–∞—Б–µ–ї–µ–љ–Є—П (–љ–∞–њ—А–Є–Љ–µ—А, 8,5% –≤ –≤—Л–±–Њ—А–Ї–µ –Є–Ј 400 —З–µ–ї–Њ–≤–µ–Ї [12]). –•–Њ—В—П —З–∞—Б—В–Њ –≥–Њ–≤–Њ—А–Є—В—Б—П, —З—В–Њ —Н—В–Є–Њ–ї–Њ–≥–Є—П –Ф–°–Ґ –Є–Љ–µ–µ—В –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–є –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В, –Є—Б—З–µ—А–њ—Л–≤–∞—О—Й–µ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞ –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ—Л—Е —А–Њ–ї–µ–є —Д–∞–Ї—В–Њ—А–Њ–≤ –Њ–Ї—А—Г–ґ–∞—О—Й–µ–є —Б—А–µ–і—Л (–њ–Є—В–∞–љ–Є–µ, —Н–Ї–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –Њ–±—Б—В–∞–љ–Њ–≤–Ї–∞, –≥–Є–≥–Є–µ–љ–∞ –і–≤–Є–ґ–µ–љ–Є—П, –њ—Б–Є—Е–Њ—Н–Љ–Њ—Ж–Є–Њ–љ–∞–ї—М–љ—Л–є —Д–Њ–љ) –Є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤ –љ–µ –њ—А–Њ–≤–Њ–і–Є–ї—Б—П. –Ъ–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–Љ–Є –і–µ—Д–µ–Ї—В–∞–Љ–Є –≤ COL11A1, COL2A1 –Є –і—А—Г–≥–Є—Е –Ї–Њ–ї–ї–∞–≥–µ–љ–∞—Е (–љ–∞–њ—А–Є–Љ–µ—А, —Б–Є–љ–і—А–Њ–Љ –°—В–Є–Ї–ї–µ—А–∞, –љ–Њ–Љ–µ—А 108300 –њ–Њ –±–∞–Ј–µ –і–∞–љ–љ—Л—Е OMIM, www.ncbi.nlm.nih.gov/omim/), —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –±–Њ–ї–µ–µ —Б–µ—А—М–µ–Ј–љ–Њ–є —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–Њ–є –Є, –Ї—А–Њ–Љ–µ —В–Њ–≥–Њ, –≤—Б—В—А–µ—З–∞—О—В—Б—П –≤–µ—Б—М–Љ–∞ —А–µ–і–Ї–Њ (–љ–µ –±–Њ–ї–µ–µ 1 —Б–ї—Г—З–∞—П –љ–∞ 10 —В—Л—Б. —З–µ–ї–Њ–≤–µ–Ї). –Ь–Њ–ґ–љ–Њ —Б–Ї–∞–Ј–∞—В—М, —З—В–Њ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –≤–Њ–≤–ї–µ—З–µ–љ–љ—Л–µ –≤ —Н—В–Є–Њ–ї–Њ–≥–Є—О –і–Є—Б–њ–ї–∞–Ј–Є–є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є, –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –љ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ—Л.
–†–∞–љ–љ—П—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞ –љ–Ф–°–Ґ, –Њ—Б–Њ–±–µ–љ–љ–Њ —Г –і–µ—В–µ–є, –њ–Њ–Ј–≤–Њ–ї—П–µ—В –Њ—Б—Г—Й–µ—Б—В–≤–ї—П—В—М —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й—Г—О —А–µ–∞–±–Є–ї–Є—В–∞—Ж–Є–Њ–љ–љ—Г—О —В–µ—А–∞–њ–Є—О –Є –њ—А–µ–і–Њ—В–≤—А–∞—Й–∞—В—М –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Ю–і–љ–Є–Љ –Є–Ј –љ–∞–Є–±–Њ–ї–µ–µ —П—А–Ї–Є—Е —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Є—Е —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ —П–≤–ї—П–µ—В—Б—П —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ–µ –ї–µ—З–µ–љ–Є–µ –і–µ—В–µ–є —Б –Ф–°–Ґ (–≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ —Б –њ—А–Њ–ї–∞–њ—Б–Њ–Љ –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞) –њ—А–Є –њ–Њ–Љ–Њ—Й–Є –Љ–∞–≥–љ–Є–є–—Б–Њ–і–µ—А–ґ–∞—Й–µ–≥–Њ –њ—А–µ–њ–∞—А–∞—В–∞ –Љ–∞–≥–љ–µ–Ј–Є—Г–Љ–Њ—А–Њ—В–∞—В–∞ (–Ь–∞–≥–љ–µ—А–Њ—В), –∞ —В–∞–Ї–ґ–µ –і—А—Г–≥–Є—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ [13]. –Р–љ–∞–ї–Є–Ј —Б–Њ–Њ—В–љ–Њ—И–µ–љ–Є—П –Љ–µ–ґ–і—Г –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–µ–є Mg2+ –Є –Ф–°–Ґ –љ–∞ —Г—А–Њ–≤–љ–µ –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е –≥–µ–љ–Њ–≤ –Љ–Њ–ґ–µ—В –њ—А–Є–≤–µ—Б—В–Є –Ї –њ–Њ–ї–µ–Ј–љ—Л–Љ –і–ї—П –њ—А–∞–Ї—В–Є–Ї—Г—О—Й–µ–≥–Њ –≤—А–∞—З–∞ –≤—Л–≤–Њ–і–∞–Љ –њ—А–Є–Љ–µ–љ–Є—В–µ–ї—М–љ–Њ –Ї –ї–µ—З–µ–љ–Є—О –Ф–°–Ґ. –Ф–∞–љ–љ—Л–є –∞–љ–∞–ї–Є–Ј —В–∞–Ї–ґ–µ –≤–∞–ґ–µ–љ –і–ї—П —Д—Г–љ–і–∞–Љ–µ–љ—В–∞–ї—М–љ–Њ–≥–Њ –њ–Њ–љ–Є–Љ–∞–љ–Є—П –њ–∞—В–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є–Є –Є —Н—В–Є–Њ–ї–Њ–≥–Є–Є –Ф–°–Ґ.
–Ґ–µ—А–Љ–Є–љ «–і–Є—Б–њ–ї–∞–Ј–Є—П» –Њ–±–Њ–Ј–љ–∞—З–∞–µ—В –∞–±–љ–Њ—А–Љ–∞–ї—М–љ—Л–є —А–Њ—Б—В/—А–∞–Ј–≤–Є—В–Є–µ —В–Ї–∞–љ–Є –Є–ї–Є –Њ—А–≥–∞–љ–∞. –Ф–Є–∞–≥–љ–Њ–Ј –Ф–°–Ґ —Б—В–∞–≤–Є—В—Б—П –љ–∞ –Њ—Б–љ–Њ–≤–µ —В—Й–∞—В–µ–ї—М–љ–Њ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞ —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ –Є —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є. –Ґ–µ–Љ –љ–µ –Љ–µ–љ–µ–µ –і–Є–∞–≥–љ–Њ–Ј –Ф–°–Ґ –љ–∞ –њ—А–∞–Ї—В–Є–Ї–µ —А–µ–і–Ї–Њ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –Ї–∞–Ї–Є–Љ–Є––ї–Є–±–Њ –Ї–Њ–љ–Ї—А–µ—В–љ—Л–Љ–Є –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є—П–Љ–Є. –°–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ –і–Є—Б–њ–ї–∞–Ј–Є—П, –Њ–±–љ–∞—А—Г–ґ–µ–љ–љ–∞—П –љ–∞ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–Љ —Г—А–Њ–≤–љ–µ, –Љ–Њ–ґ–µ—В —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Њ–≤–∞—В—М –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ –≤ —Б—В—А—Г–Ї—В—Г—А–µ —В–Ї–∞–љ–Є.
–Т —Б–ї—Г—З–∞–µ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –і–Є—Б–њ–ї–∞–Ј–Є—П (—В.–µ. «–∞–±–љ–Њ—А–Љ–∞–ї—М–љ—Л–є —А–Њ—Б—В») –Љ–Њ–ґ–µ—В –њ—А–Њ–Є—Б—Е–Њ–і–Є—В—М –≤—Б–ї–µ–і—Б—В–≤–Є–µ: 1) –∞–±–љ–Њ—А–Љ–∞–ї—М–љ–Њ–≥–Њ —Б–Є–љ—В–µ–Ј–∞ –Є–ї–Є —Б–±–Њ—А–Ї–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞; 2) —Б–Є–љ—В–µ–Ј–∞ –∞–±–љ–Њ—А–Љ–∞–ї—М–љ–Њ–≥–Њ –Ї–Њ–ї–ї–∞–≥–µ–љ–∞; 3) —З—А–µ–Ј–Љ–µ—А–љ–Њ–є –і–µ–≥—А–∞–і–∞—Ж–Є–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞; 4) –љ–∞—А—Г—И–µ–љ–Є–є —Б—В—А—Г–Ї—В—Г—А—Л –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ, –≤—Б–ї–µ–і—Б—В–≤–Є–µ –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ–є –њ–Њ–њ–µ—А–µ—З–љ–Њ–є —Б—И–Є–≤–Ї–Є; 5) –∞–љ–∞–ї–Њ–≥–Є—З–љ—Л—Е –∞–љ–Њ–Љ–∞–ї–Є–є, —Б–≤—П–Ј–∞–љ–љ—Л—Е —Б —Н–ї–∞—Б—В–Є–љ–Њ–≤—Л–Љ–Є –≤–Њ–ї–Њ–Ї–љ–∞–Љ–Є; 6) —А–∞–Ј—А—Г—И–µ–љ–Є—П —В–Ї–∞–љ–Є –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ –∞—Г—В–Њ–Є–Љ—Г–љ–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є; 7) –Љ–љ–Њ–≥–Є—Е –і—А—Г–≥–Є—Е, –љ–µ –Є–Ј—Г—З–µ–љ–љ—Л—Е –љ–∞ —Б–µ–≥–Њ–і–љ—П—И–љ–Є–є –Љ–Њ–Љ–µ–љ—В –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤.
–Ф–∞–ї–µ–µ –Љ—Л —Б–Є—Б—В–µ–Љ–∞—В–Є—З–µ—Б–Ї–Є —А–∞—Б—Б–Љ–Њ—В—А–Є–Љ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ––Ї–ї–µ—В–Њ—З–љ—Г—О –±–Є–Њ–ї–Њ–≥–Є—О —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є, –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ –Љ–∞–≥–љ–Є—П, –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–є –Љ–∞–≥–љ–Є—П –Є —Б–Њ—Б—В–Њ—П–љ–Є—П –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є.
–Ь–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ––Ї–ї–µ—В–Њ—З–љ–∞—П –±–Є–Њ–ї–Њ–≥–Є—П
—Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є
–Ґ–µ—А–Љ–Є–љ «—Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–∞—П —В–Ї–∞–љ—М» –њ—А–Є–љ–∞–і–ї–µ–ґ–Є—В –≤ –Њ—Б–љ–Њ–≤–љ–Њ–Љ –Ї –Њ–±–ї–∞—Б—В–Є —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є–Є –Є –±–Є–Њ–Љ–µ–і–Є—Ж–Є–љ—Л. –Ф–ї—П —В–Њ–≥–Њ —З—В–Њ–±—Л –њ—А–Є–±–ї–Є–Ј–Є—В—М—Б—П –Ї –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л–Љ –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞–Љ, –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–≥—Г—В –±—Л—В—М –≤–Њ–≤–ї–µ—З–µ–љ—Л –≤ –Ф–°–Ґ, –Љ—Л –і–Њ–ї–ґ–љ—Л –Њ–њ–µ—А–Є—А–Њ–≤–∞—В—М —В–µ—А–Љ–Є–љ–∞–Љ–Є –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ–є –±–Є–Њ–ї–Њ–≥–Є–Є –Ї–ї–µ—В–Ї–Є, —В–∞–Ї–Є–Љ–Є –Ї–∞–Ї «–≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–∞—П –Љ–∞—В—А–Є—Ж–∞», «–Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ–∞—П –∞–і–≥–µ–Ј–Є—П», «–∞–і–≥–µ–Ј–Є—П –Љ–∞—В—А–Є—Ж–∞––Ї–ї–µ—В–Ї–∞», «–њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ—Л» –Є –і—А.
–°–ї–Њ–ґ–љ–Њ—Б—В—М –ї—О–±–Њ–є —В–Ї–∞–љ–Є —З–µ–ї–Њ–≤–µ—З–µ—Б–Ї–Њ–≥–Њ —В–µ–ї–∞ –њ—А–Є —А–∞—Б—Б–Љ–Њ—В—А–µ–љ–Є–Є –љ–∞ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ–Љ —Г—А–Њ–≤–љ–µ –њ—А–Њ—Б—В–Њ –Њ—И–µ–ї–Њ–Љ–ї—П–µ—В, –Є –≤ —Н—В–Њ–Љ –Њ—В–љ–Њ—И–µ–љ–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–∞—П —В–Ї–∞–љ—М –≤–Њ–≤—Б–µ –љ–µ –Є—Б–Ї–ї—О—З–µ–љ–Є–µ. –І–∞—Б—В–Њ –≥–Њ–≤–Њ—А–Є—В—Б—П, —З—В–Њ –Њ—Б–љ–Њ–≤–љ—Л–µ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В—Л —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є – —Н—В–Њ –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞, –≥–Є–±–Ї–Є–µ –≤–Њ–ї–Њ–Ї–љ–∞, –Ї–ї–µ—В–Ї–Є –Є —А–µ–Љ–Њ–і–µ–ї–Є—А—Г—О—Й–Є–µ —Д–µ—А–Љ–µ–љ—В—Л. –Э–Њ —В–∞–Ї–Њ–≥–Њ —А–Њ–і–∞ –Њ–њ–Є—Б–∞–љ–Є—П –љ–µ –Њ—В—А–∞–ґ–∞—О—В —Д–∞–Ї—В–Є—З–µ—Б–Ї—Г—О —Б–ї–Њ–ґ–љ–Њ—Б—В—М —В–Ї–∞–љ–Є –љ–∞ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ–Љ —Г—А–Њ–≤–љ–µ –Є —А–µ–∞–ї—М–љ–Њ–µ —З–Є—Б–ї–Њ –≤–Њ–≤–ї–µ—З–µ–љ–љ—Л—Е –≥–µ–љ–Њ–≤. –Я–Њ—Н—В–Њ–Љ—Г –і–∞–љ–љ—Л–є —А–∞–Ј–і–µ–ї –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є —Б–ґ–∞—В–Њ–µ —А–µ–Ј—О–Љ–µ —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –Љ–∞–Ї—А–Њ–Љ–Њ–ї–µ–Ї—Г–ї, –ї–µ–ґ–∞—Й–Є—Е –≤ –Њ—Б–љ–Њ–≤–µ —Г–ї—М—В—А–∞—Б—В—А—Г–Ї—В—Г—А—Л —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є —Б —Ж–µ–ї—М—О —Д–Њ—А–Љ—Г–ї–Є—А–Њ–≤–Ї–Є –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е –Є–љ—В–µ—А–њ—А–µ—В–∞—Ж–Є–є –Ф–°–Ґ.
–Я—А–Є–љ—Ж–Є–њ–Є–∞–ї—М–љ–Њ–µ –Њ—В–ї–Є—З–Є–µ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –Њ—В –ї—О–±–Њ–≥–Њ –і—А—Г–≥–Њ–≥–Њ —В–Є–њ–∞ —В–Ї–∞–љ–Є – —Н—В–Њ –Є–Ј–±—Л—В–Њ–Ї –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж—Л –њ—А–Є —Б—А–∞–≤–љ–Є—В–µ–ї—М–љ–Њ –љ–µ–±–Њ–ї—М—И–Њ–Љ —З–Є—Б–ї–µ –Ї–ї–µ—В–Њ–Ї, —Б–Њ—Б—В–∞–≤–ї—П—О—Й–Є—Е —В–Ї–∞–љ—М. –Т –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ–є –±–Є–Њ–ї–Њ–≥–Є–Є –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–∞—П –Љ–∞—В—А–Є—Ж–∞ (–Т–Ъ–Ь) –Њ–њ—А–µ–і–µ–ї–µ–љ–∞, –Ї–∞–Ї —Б–ї–Њ–ґ–љ–∞—П —Б–µ—В—М, —Б—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–љ–∞—П –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–Љ–Є —Б—В—А—Г–Ї—В—Г—А–љ—Л–Љ–Є –Љ–∞–Ї—А–Њ–Љ–Њ–ї–µ–Ї—Г–ї–∞–Љ–Є (—В–∞–Ї–Є–Љ–Є –Ї–∞–Ї –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ—Л, –Ї–Њ–ї–ї–∞–≥–µ–љ—Л, –Є —Н–ї–∞—Б—В–Є–љ). –Т–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г—П –і—А—Г–≥ —Б –і—А—Г–≥–Њ–Љ –Є —Б –Ї–ї–µ—В–Ї–∞–Љ–Є, —Н—В–Є —Б—В—А—Г–Ї—В—Г—А–љ—Л–µ –Љ–∞–Ї—А–Њ–Љ–Њ–ї–µ–Ї—Г–ї—Л –њ–Њ–і–і–µ—А–ґ–Є–≤–∞—О—В —Б—В—А—Г–Ї—В—Г—А–љ—Г—О —Ж–µ–ї–Њ—Б—В–љ–Њ—Б—В—М —В–Ї–∞–љ–µ–є [14]. –Ш–Љ–µ–љ–љ–Њ –Љ–∞—В—А–Є—Ж–∞ –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–µ—В –Њ—А–≥–∞–љ–Є–Ј–Њ–≤–∞–љ–љ—Г—О —Б—А–µ–і—Г, –≤ –њ—А–µ–і–µ–ї–∞—Е –Ї–Њ—В–Њ—А–Њ–є –Љ–Є–≥—А–Є—А—Г—О—Й–Є–µ –Ї–ї–µ—В–Ї–Є –Љ–Њ–≥—Г—В –њ–µ—А–µ–Љ–µ—Й–∞—В—М—Б—П –Є –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Њ–≤–∞—В—М –і—А—Г–≥ —Б –і—А—Г–≥–Њ–Љ. –Т—Б–µ –Љ–∞–Ї—А–Њ–Љ–Њ–ї–µ–Ї—Г–ї—Л, —Б–Њ—Б—В–∞–≤–ї—П—О—Й–Є–µ –Т–Ъ–Ь, –њ—А–Њ–Є–Ј–≤–Њ–і—П—В—Б—П –Ї–ї–µ—В–Ї–∞–Љ–Є –≤ –Љ–∞—В—А–Є—Ж–µ. –Т –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ—Л—Е —В–Ї–∞–љ–µ–є –Љ–∞—В—А–Є—З–љ—Л–µ –Љ–∞–Ї—А–Њ–Љ–Њ–ї–µ–Ї—Г–ї—Л —Б–Є–љ—В–µ–Ј–Є—А—Г—О—В—Б—П —Д–Є–±—А–Њ–±–ї–∞—Б—В–∞–Љ–Є, –∞ –≤ —Б–њ–µ—Ж–Є–∞–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е —В–Є–њ–∞—Е —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є, —В–∞–Ї–Є—Е –Ї–∞–Ї, –љ–∞–њ—А–Є–Љ–µ—А, —Е—А—П—Й –Є –Ї–Њ—Б—В—М – —Е–Њ–љ–і—А–Њ–±–ї–∞—Б—В–∞–Љ–Є –Є –Њ—Б—В–µ–Њ–±–ї–∞—Б—В–∞–Љ–Є. –Т–Ъ–Ь —Б–Њ—Б—В–Њ–Є—В –Є–Ј —В—А–µ—Е –њ—А–Є–љ—Ж–Є–њ–Є–∞–ї—М–љ—Л—Е –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤: –≥–µ–ї–µ–Њ–±—А–∞–Ј–љ–Њ–є —Б—А–µ–і—Л, –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ –Є —Н–ї–∞—Б—В–Є–љ–Њ–≤—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ.
–У–µ–ї–µ–Њ–±—А–∞–Ј–љ–∞—П —Б—А–µ–і–∞. –Э–∞–Є–≤–∞–ґ–љ–µ–є—И–Є–є –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж—Л – —Н—В–Њ –≥–µ–ї–µ–Њ–±—А–∞–Ј–љ–∞—П —Б—А–µ–і–∞, —Д–Њ—А–Љ–Є—А—Г–µ–Љ–∞—П –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ–∞–Љ–Є: —З—А–µ–Ј–≤—Л—З–∞–є–љ–Њ —А–∞—Б—В—П–љ—Г—В—Л–Љ–Є –њ–Њ–ї–Є–њ–µ–њ—В–Є–і–љ—Л–Љ–Є —Ж–µ–њ—П–Љ–Є —Б –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–Љ–Є –њ–Њ–ї–Є—Б–∞—Е–∞—А–Є–і–љ—Л–Љ–Є —Ж–µ–њ—П–Љ–Є –≥–ї—О–Ї–Њ–Ј–∞–Љ–Є–љ–Њ–≥–ї–Є–Ї–∞–љ–Њ–≤, –њ—А–Є—Б–Њ–µ–і–Є–љ–µ–љ–љ—Л—Е –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ –Ї–Њ–≤–∞–ї–µ—В–љ—Л—Е —Б–≤—П–Ј–µ–є (—А–Є—Б. 1).
–Ь–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–µ —Ж–µ–њ–Є –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ–Њ–≤ –њ—А–Є–Ї—А–µ–њ–ї—П—О—В—Б—П –Ї –Њ—Б–Њ–±–Њ–Љ—Г –≤–Є–і—Г –≥–ї—О–Ї–Њ–Ј–∞–Љ–Є–љ–Њ–≥–ї–Є–Ї–∞–љ–∞ – –њ–Њ–ї–Є–Љ–µ—А—Г –≥–Є–∞–ї—Г—А–Њ–љ–Њ–≤–Њ–є –Ї–Є—Б–ї–Њ—В—Л, –љ–∞–Ј—Л–≤–∞–µ–Љ–Њ–Љ—Г –≥–Є–∞–ї—Г—А–Њ–љ–∞–љ–Њ–Љ. –Э–Є—В–Є –≥–Є–∞–ї—Г—А–Њ–љ–∞–љ–∞ —Б–Ї—А–µ–њ–ї—П—О—В —Б—В—А—Г–Ї—В—Г—А—Г –≥–µ–ї—П –≤ –µ–і–Є–љ–Њ–µ —Ж–µ–ї–Њ–µ, –Є —Н—В–Њ—В –њ–Њ–ї–Є—Б–∞—Е–∞—А–Є–і–љ—Л–є «–≥–µ–ї—М» –Љ–Њ–ґ–µ—В –њ—А–Њ—В–Є–≤–Њ—Б—В–Њ—П—В—М —Б–ґ–∞—В–Є—О –Є —А–∞—Б—В—П–ґ–µ–љ–Є—О –Т–Ъ–Ь –Є –≤ —В–Њ –ґ–µ –≤—А–µ–Љ—П –Њ–±–µ—Б–њ–µ—З–Є–≤–∞—В—М –±—Л—Б—В—А—Г—О –і–Є—Д—Д—Г–Ј–Є—О –њ–Є—В–∞—В–µ–ї—М–љ—Л—Е –≤–µ—Й–µ—Б—В–≤, —Б—В—А–Њ–Є—В–µ–ї—М–љ—Л—Е –Љ–∞—В–µ—А–Є–∞–ї–Њ–≤ –Є –≥–Њ—А–Љ–Њ–љ–Њ–≤ –Љ–µ–ґ–і—Г –Ї—А–Њ–≤—М—О –Є –Ї–ї–µ—В–Ї–∞–Љ–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є.
–Ь–µ—Е–∞–љ–Є—З–µ—Б–Ї–Є —Б—В—А—Г–Ї—В—Г—А–∞ –≥–µ–ї—П —Г—Б–Є–ї–µ–љ–∞ –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ –≤–Њ–ї–Њ–Ї–Њ–љ —В—А–µ—Е –Њ—Б–љ–Њ–≤–љ—Л—Е —В–Є–њ–Њ–≤: 1) –Ї–Њ–ї–ї–∞–≥–µ–љo–≤—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ (—Б–Њ—Б—В–Њ—П—Й–Є—Е –≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ –Є–Ј –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ I), –Ї–Њ—В–Њ—А—Л–µ —Д–Њ—А–Љ–Є—А—Г—О—В —Б–Ї–µ–ї–µ—В —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є; 2) –≥–Є–±–Ї–Є—Е –≤–Њ–ї–Њ–Ї–Њ–љ (—Б–Њ—Б—В–Њ—П—Й–Є—Е –≤ –Њ—Б–љ–Њ–≤–љ–Њ–Љ –Є–Ј —Н–ї–∞—Б—В–Є–љ–∞ –Є —Д–Є–±—А–Є–ї–ї–Є–љ–Њ–≤), –Ї–Њ—В–Њ—А—Л–µ –њ—А–Є–і–∞—О—В —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є —Н–ї–∞—Б—В–Є—З–љ–Њ—Б—В—М; 3) —Б–µ—В—З–∞—В—Л—Е (–Є–ї–Є —А–µ—В–Є–Ї—Г–ї—П—А–љ—Л—Е) –≤–Њ–ї–Њ–Ї–Њ–љ (–Ї–Њ–ї–ї–∞–≥–µ–љ III), –Ї–Њ—В–Њ—А—Л–µ –Њ–±—А–∞–Ј—Г—О—В –њ–µ—А–µ–Ї—А–µ—Б—В–љ—Л–µ —Б–≤—П–Ј–Є –Љ–µ–ґ–і—Г –≤—Б–µ–Љ–Є –і—А—Г–≥–Є–Љ–Є –≤–Њ–ї–Њ–Ї–љ–∞–Љ–Є –Є –і–µ—А–ґ–∞—В –≤–Љ–µ—Б—В–µ –≤—Б–µ –Њ—Б—В–∞–ї—М–љ—Л–µ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В—Л —В–Ї–∞–љ–Є.
–У–µ–ї–µ–Њ–±—А–∞–Ј–љ–∞—П —Б—Г–±—Б—В–∞–љ—Ж–Є—П –Т–Ъ–Ь —Б—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–∞ –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ–∞–Љ–Є –Є –Љ–љ–Њ–≥–Њ–і–Њ–Љ–µ–љ–љ—Л–Љ–Є –≥–ї–Є–Ї–Њ–±–µ–ї–Ї–∞–Љ–Є. –Я—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ—Л –њ—А–Є–Ї—А–µ–њ–ї—П—О—В—Б—П –Ї –љ–Є—В—П–Љ –≥–Є–∞–ї—Г—А–Њ–љ–∞–љ–∞, –Ї–∞–ґ–і–∞—П –Є–Ј –Ї–Њ—В–Њ—А—Л—Е —Б–Њ–і–µ—А–ґ–Є—В –±–Њ–ї–µ–µ 25 —В—Л—Б. –Љ–Њ–љ–Њ–Љ–µ—А–Њ–≤ –≥–Є–∞–ї—Г—А–Њ–љ–Њ–≤–Њ–є –Ї–Є—Б–ї–Њ—В—Л, –Ї–∞–ґ–і–∞—П –љ–Є—В—М –Љ–Њ–ґ–µ—В –Є–Љ–µ—В—М –і–ї–Є–љ—Г –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ –і–µ—Б—П—В–Ї–Њ–≤ –Љ–Є–Ї—А–Њ–љ. –У–Є–∞–ї—Г—А–Њ–љ–∞–љ —Б–Є–љ—В–µ–Ј–Є—А—Г–µ—В—Б—П –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ –≥–Є–∞–ї—Г—А–Њ–љ–∞–љ—Б–Є–љ—В–µ—В–∞–Ј (–≥–µ–љ—Л HAS1, HAS2 –Є HAS3) –Є –і–µ–≥—А–∞–і–Є—А—Г–µ—В—Б—П –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ –≥–Є–∞–ї—Г—А–Њ–љ–Є–і–∞–Ј (–≥–µ–љ—Л HYAL2, HYAL3, HYAL4 –Є HYALP).
–Я—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ—Л —Б–Њ–і–µ—А–ґ–∞—В –і–ї–Є–љ–љ—Л–µ —Ж–µ–њ–Њ—З–Ї–Є –њ–Њ–ї–Є—Б–∞—Е–∞—А–Є–і–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ –њ—А–Є —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Г—Б–ї–Њ–≤–Є—П—Е –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ–Њ –Ј–∞—А—П–ґ–µ–љ—Л –≤—Б–ї–µ–і—Б—В–≤–Є–µ –Ї–Њ–≤–∞–ї–µ–љ—В–љ–Њ –њ—А–Є—Б–Њ–µ–і–Є–љ–µ–љ–љ—Л—Е —Б—Г–ї—М—Д–∞—В–Њ–≤ –Є —Г—А–Њ–љ–∞—В–Њ–≤. –Ф–ї—П –Ї–∞–ґ–і–Њ–≥–Њ —В–Є–њ–∞ –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ–∞ –µ—Б—В—М –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–µ –±–µ–ї–Ї–Є, –Ї–Њ—В–Њ—А—Л–µ —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є —Б–≤—П–Ј—Л–≤–∞—О—В—Б—П —Б —Н—В–Є–Љ –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ–Њ–Љ, –∞ —В–∞–Ї–ґ–µ –љ–µ –Љ–µ–љ–µ–µ –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–µ —Б–Є–љ—В–µ—В–∞–Ј—Л, –≤–Њ–≤–ї–µ—З–µ–љ–љ—Л–µ –≤ —Б–Є–љ—В–µ–Ј —Ж–µ–њ–µ–є –≥–ї—О–Ї–Њ–Ј–∞–Љ–Є–љ–Њ–≥–ї–Є–Ї–∞–љ–Њ–≤ –Є –Є—Е –њ—А–Є—Б–Њ–µ–і–Є–љ–µ–љ–Є–µ –≤ –±–µ–ї–Ї–Њ–≤–Њ–є —Б–µ—А–і—Ж–µ–≤–Є–љ–µ. –Я—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ—Л –Є —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–Є–µ –≥–µ–љ—Л –Ї–ї–∞—Б—Б–Є—Д–Є—Ж–Є—А—Г—О—В—Б—П —Б–Њ–≥–ї–∞—Б–љ–Њ –Є—Е –≥–ї—О–Ї–Њ–Ј–∞–Љ–Є–љ–Њ–≥–ї–Є–Ї–∞–љ–љ—Л–Љ —Ж–µ–њ—П–Љ, –Є –Њ—Б–љ–Њ–≤–љ—Л–µ —В–Є–њ—Л –≤–Ї–ї—О—З–∞—О—В —Е–Њ–љ–і—А–Њ–Є—В–Є–љ—Б—Г–ї—М—Д–∞—В –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ (–≥–µ–љ—Л CSPG1, CSPG2, CSPG3, CSPG4, CSPG5, CSPG6) –Є –≥–µ–њ–∞—А–∞–љ—Б—Г–ї—М—Д–∞—В –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ (–њ–µ—А–ї–µ–Ї–∞–љ, –≥–µ–љ HSPG2). –≠—В–Є –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ—Л –≤–Њ–≤–ї–µ—З–µ–љ—Л –≤ –Њ—Б–љ–Њ–≤–љ–Њ–Љ –≤ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Њ—Б–љ–Њ–≤–љ–Њ–є —Б—В—А—Г–Ї—В—Г—А—Л –≥–µ–ї—П. –Ь—Г—В–∞—Ж–Є–Є –≤ —Е–Њ–љ–і—А–Њ–Є—В–Є–љ—Б—Г–ї—М—Д–∞—В –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ–∞—Е –Љ–Њ–≥—Г—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї —Б–Ї–µ–ї–µ—В–љ–Њ–є –і–Є—Б–њ–ї–∞–Ј–Є–Є [15]. –У–µ–њ–∞—А–∞–љ—Б—Г–ї—М—Д–∞—В –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ —Г—З–∞—Б—В–≤—Г–µ—В –≤ –Ї–ї–µ—В–Њ—З–љ–Њ–є –∞–і–≥–µ–Ј–Є–Є, –Њ–±–ї–∞–і–∞–µ—В –∞–љ–≥–Є–Њ–≥–µ–љ–љ—Л–Љ–Є —Б–≤–Њ–є—Б—В–≤–∞–Љ–Є –Є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –і–µ—Д–µ–Ї—В—Л –≤ –≥–µ–љ–µ –њ–µ—А–ї–µ–Ї–∞–љ–∞ –њ—А–Є–≤–Њ–і—П—В –Ї —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –Љ–Є–Њ—В–Њ–љ–Є–Є –Є —Б–Ї–µ–ї–µ—В–љ–Њ–є –і–Є—Б–њ–ї–∞–Ј–Є–Є [16]. –Ф–µ–Ї–Њ—А–Є–љ (–≥–µ–љ DCN) –Є –ї—О–Љ–Є–Ї–∞–љ (–≥–µ–љ LUM) –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г—О—В —Б –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–Љ–Є –≤–Њ–ї–Њ–Ї–љ–∞–Љ–Є –Є –Њ–≥—А–∞–љ–Є—З–Є–≤–∞—О—В –і–Є–∞–Љ–µ—В—А –≤–Њ–ї–Њ–Ї–Њ–љ (—Б–Љ. –љ–Є–ґ–µ). –Ъ–µ—А–∞—В–∞–љ—Б—Г–ї—М—Д–∞—В–љ—Л–є –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ –ї—О–Љ–Є–Ї–∞–љ –≤—Б—В—А–µ—З–∞–µ—В—Б—П –≤ —А–Њ–≥–Њ–≤–Є—Ж–µ –Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л—Е –Т–Ъ–Ь —Б–µ—А–і—Ж–∞, –∞–Њ—А—В—Л, —Б–Ї–µ–ї–µ—В–љ–Њ–є –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А—Л, –Ї–Њ–ґ–Є –Є –Љ–µ–ґ–њ–Њ–Ј–≤–Њ–љ–Њ—З–љ—Л—Е –і–Є—Б–Ї–Њ–≤. –Ґ—А–∞–љ—Б–≥–µ–љ–љ—Л–µ –Љ—Л—И–Є —Б –і–µ—Д–Є—Ж–Є—В–Њ–Љ –ї—О–Љ–Є–Ї–∞–љ–∞ –і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–ї–Є —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –≤ –ґ–µ—Б—В–Ї–Њ—Б—В–Є —Б—Г—Е–Њ–ґ–Є–ї–Є–є, –њ–Њ–≤—Л—И–µ–љ–љ—Г—О —Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М –Ї –≤—Л–≤–Є—Е–∞–Љ —Б—Г—Б—В–∞–≤–Њ–≤ –Є –Њ—Б—В–µ–Њ–∞—А—В—А–Є—В [17].
–§–µ—А–Љ–µ–љ—В—Л, —Г—З–∞—Б—В–≤—Г—О—Й–Є–µ –≤ –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є—Е –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є—П—Е –Є –њ—А–Є—Б–Њ–µ–і–Є–љ–µ–љ–Є–Є –≥–ї—О–Ї–Њ–Ј–∞–Љ–Є–љ–Њ–≥–ї–Є–Ї–∞–љ–Њ–≤, –Љ–Њ–≥—Г—В –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –≤–ї–Є—П—В—М –љ–∞ —Б—В—А—Г–Ї—В—Г—А—Г –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж—Л. –Э–∞–њ—А–Є–Љ–µ—А, –і–µ—Д–Є—Ж–Є—В –Ї—Б–Є–ї–Њ–Ј–Є–ї b–1,4––≥–∞–ї–∞–Ї—В–Њ–Ј–Є–ї—В—А–∞–љ—Б—Д–µ—А–∞–Ј—Л 7 (–≥–µ–љ B4GALT7) —Б–≤—П–Ј–∞–љ —Б –Њ–і–љ–Њ–є –Є–Ј —Д–Њ—А–Љ —Б–Є–љ–і—А–Њ–Љ–∞ –≠–ї–µ—А—Б–∞––Ф–∞–љ–ї–Њ (—Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М –Ї –≤—Л–≤–Є—Е–∞–Љ, —Е—А—Г–њ–Ї–∞—П –Є–ї–Є –≥–Є–њ–µ—А—Н–ї–∞—Б—В–Є—З–љ–∞—П –Ї–Њ–ґ–∞, —Е—А—Г–њ–Ї–Є–µ –Ї—А–Њ–≤—П–љ—Л–µ —Б–Њ—Б—Г–і—Л –Є —В.–і., –љ–Њ–Љ–µ—А –њ–Њ OMIM 130000) [18].
–Ь–љ–Њ–≥–Њ–і–Њ–Љ–µ–љ–љ—Л–µ –≥–ї–Є–Ї–Њ–±–µ–ї–Ї–Є –≤–Ї–ї—О—З–∞—О—В —Д–Є–±—А–Њ–љ–µ–Ї—В–Є–љ, –ї–∞–Љ–Є–љ–Є–љ—Л –Є —В–µ–љ–∞—Б—Ж–Є–љ—Л. –Ф–∞–љ–љ—Л–µ –±–µ–ї–Ї–Є —Б–Њ—Б—В–Њ—П—В –Є–Ј –і–µ—Б—П—В–Ї–Њ–≤ –Њ–і–љ–Њ—А–Њ–і–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –Љ–Њ–і—Г–ї–µ–є (–і–Њ–Љ–µ–љ–Њ–≤), –Ї–Њ–≤–∞–ї–µ–љ—В–љ–Њ —Б–≤—П–Ј–∞–љ–љ—Л—Е –і—А—Г–≥ —Б –і—А—Г–≥–Њ–Љ —Б –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ–Љ —Ж–µ–њ–µ–є. –§–Є–±—А–Њ–љ–µ–Ї—В–Є–љ (–≥–µ–љ FN1) —Г—З–∞—Б—В–≤—Г–µ—В –≤ –∞–і–≥–µ–Ј–Є–Є –Ї–ї–µ—В–Њ–Ї –Ї –Т–Ъ–Ь, –∞ —В–∞–Ї–ґ–µ –≤ –њ—А–Њ—Ж–µ—Б—Б–∞—Е –Љ–Є–≥—А–∞—Ж–Є–Є –Ї–ї–µ—В–Њ–Ї, –Ј–∞–ґ–Є–≤–ї–µ–љ–Є—П —А–∞–љ, —Б–≤–µ—А—В—Л–≤–∞–љ–Є—П –Ї—А–Њ–≤–Є –Є –Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –Њ—В–≤–µ—В–∞. –Ы–∞–Љ–Є–љ–Є–љ—Л (–≥–µ–љ—Л LAMA1, LAMA2, LAMA3, LAMA4; LAMB1, LAMB2; LAMC1, LAM–°2) –≤–∞–ґ–љ—Л –і–ї—П –і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–∞–љ–Є—П, –∞–і–≥–µ–Ј–Є–Є –Є –Љ–Є–≥—А–∞—Ж–Є–Є –Ї–ї–µ—В–Њ–Ї; –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –і–µ—Д–µ–Ї—В—Л –≤ –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ –ї–∞–Љ–Є–љ–Є–љ–Њ–≤ –њ—А–Є–≤–Њ–і—П—В –Ї —Б–µ—А—М–µ–Ј–љ—Л–Љ —Д–Њ—А–Љ–∞–Љ –Љ—Г—Б–Ї—Г–ї—М–љ–Њ–є –і–Є—Б—В—А–Њ—Д–Є–Є –Є –±—Г–ї–µ–Ј–љ–Њ–≥–Њ —Н–њ–Є–і–µ—А–Љ–Њ–ї–Є–Ј–∞. –Ґ–µ–љ–∞—Б—Ж–Є–љ—Л (TNXB, TNC), –≤–µ—А–Њ—П—В–љ–Њ, –Є–≥—А–∞—О—В —Б—В—А—Г–Ї—В—Г—А–љ—Г—О —А–Њ–ї—М, –Є –і–µ—Д–µ–Ї—В—Л –≤ —В–µ–љ–∞—Б—Ж–Є–љ–∞—Е –Љ–Њ–≥—Г—В –њ—А–Њ–≤–Њ–і–Є—В—М –Ї —Б–Є–љ–і—А–Њ–Љ—Г –≠–ї–µ—А—Б–∞––Ф–∞–љ–ї–Њ [19].
–Ъ–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞ –њ—А–Є–і–∞—О—В —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –њ—А–Њ—З–љ–Њ—Б—В—М –Є –і–Њ–ї–≥–Њ–≤–µ—З–љ–Њ—Б—В—М. –Ъ–∞–ґ–і–Њ–µ –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤–Њ–µ –≤–Њ–ї–Њ–Ї–љ–Њ –Є–Љ–µ–µ—В –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ –Љ–Є–Ї—А–Њ–Љ–µ—В—А–Њ–≤ –≤ –і–Є–∞–Љ–µ—В—А–µ –Є —Б–Њ—Б—В–Њ–Є—В –Є–Ј —В—Л—Б—П—З –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ—Л—Е –њ–Њ–ї–Є–њ–µ–њ—В–Є–і–љ—Л—Е —Ж–µ–њ–µ–є –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤, –њ–ї–Њ—В–љ–Њ —Г–њ–∞–Ї–Њ–≤–∞–љ–љ—Л—Е –≤–Љ–µ—Б—В–µ. –Ъ–Њ–ї–ї–∞–≥–µ–љ—Л (—А–Є—Б. 2) – –Њ–і–љ–Є –Є–Ј –љ–∞–Є–±–Њ–ї–µ–µ –Њ–±–Є–ї—М–љ—Л—Е –±–µ–ї–Ї–Њ–≤ –≤–Њ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж–µ –Є –≤ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Т –≥–µ–љ–Њ–Љ–µ —З–µ–ї–Њ–≤–µ–Ї–∞ —Б—Г—Й–µ—Б—В–≤—Г–µ—В –Њ–Ї–Њ–ї–Њ 50 –≥–µ–љ–Њ–≤, –Ї–Њ–і–Є—А—Г—О—Й–Є—Е —А–∞–Ј–ї–Є—З–љ—Л–µ –Ї–Њ–ї–ї–∞–≥–µ–љ—Л, –Є –њ—А–Њ–і—Г–Ї—В—Л —Н—В–Є—Е –≥–µ–љ–Њ–≤ –Њ–±—А–∞–Ј—Г—О—В –±–Њ–ї–µ–µ —З–µ–Љ 20 —В–Є–њ–Њ–≤ –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ, –љ–∞–є–і–µ–љ–љ—Л—Е –≤ —Б–∞–Љ—Л—Е —А–∞–Ј–ї–Є—З–љ—Л—Е —В–Ї–∞–љ—П—Е.
–Ъ–Њ–ї–ї–∞–≥–µ–љ—Л —А–∞–Ј–ї–Є—З–∞—О—В—Б—П –њ–Њ –Є—Е –њ–Њ–ї–Њ–ґ–µ–љ–Є—О –≤ —В–Ї–∞–љ–Є –Є –њ–Њ —Б–≤–Њ–µ–Љ—Г —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–Љ—Г –Ј–љ–∞—З–µ–љ–Є—О. –І–µ—В—Л—А–µ –Њ—Б–љ–Њ–≤–љ—Л—Е —В–Є–њ–∞ –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤ (I–IV) –≤–Ї–ї—О—З–∞—О—В —Б–ї–µ–і—Г—О—Й–Є–µ –≥–µ–љ—Л: –Ї–Њ–ї–ї–∞–≥–µ–љ I (–≥–µ–љ—Л COL1A1, COL1A2) – –Њ—Б–љ–Њ–≤–љ–Њ–є –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В –Ї–Њ—Б—В–Є, –Ї–Њ—В–Њ—А—Л–є —В–∞–Ї–ґ–µ –њ—А–Є—Б—Г—В—Б—В–≤—Г–µ—В –≤ —И—А–∞–Љ–∞—Е, —Б—Г—Е–Њ–ґ–Є–ї–Є—П—Е –Є —Е—А—П—Й–∞—Е; –Ї–Њ–ї–ї–∞–≥–µ–љ II (–≥–µ–љ COL2A1) – –Њ—Б–љ–Њ–≤–љ–Њ–є –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В —Е—А—П—Й–∞; –Ї–Њ–ї–ї–∞–≥–µ–љ III (–≥–µ–љ COL3A1) —Д–Њ—А–Љ–Є—А—Г–µ—В —А–µ—В–Є–Ї—Г–ї—П—А–љ—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞, –Ї–Њ—В–Њ—А—Л–µ –і–µ—А–ґ–∞—В –≤–Љ–µ—Б—В–µ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ—Г—О –Љ–∞—В—А–Є—Ж—Г; –Ї–Њ–ї–ї–∞–≥–µ–љ IV (–≥–µ–љ—Л COL4A1, COL4A2, COL4A3, COL4A4, COL4A5, COL4A6) —Д–Њ—А–Љ–Є—А—Г–µ—В –±–∞–Ј–∞–ї—М–љ—Г—О –ї–∞–Љ–Є–љ—Г, –љ–∞ –Ї–Њ—В–Њ—А–Њ–є –і–µ—А–ґ–Є—В—Б—П —Н–њ–Є—В–µ–ї–Є–є. –£–љ–∞—Б–ї–µ–і–Њ–≤–∞–љ–љ—Л–µ –Є —А–µ–і–Ї–Њ –≤—Б—В—А–µ—З–∞—О—Й–Є–µ—Б—П –Љ—Г—В–∞—Ж–Є–Є –≤ —Н—В–Є—Е –Є –і—А—Г–≥–Є—Е –Ї–Њ–ї–ї–∞–≥–µ–љ–∞—Е –≤ –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ –њ—А–Є–≤–Њ–і—П—В –Ї –±–Њ–ї–µ–Ј–љ–Є –≠–ї–µ—А—Б–∞––Ф–∞–љ–ї–Њ –Є –±—Г–ї–µ–Ј–љ–Њ–Љ—Г —Н–њ–Є–і–µ—А–Љ–Њ–ї–Є–Ј—Г.
–Ъ–Њ–ї–ї–∞–≥–µ–љ—Л–µ –±–Њ–ї–µ–Ј–љ–Є –Љ–Њ–≥—Г—В –≤–Њ–Ј–љ–Є–Ї–∞—В—М –љ–µ —В–Њ–ї—М–Ї–Њ –≤—Б–ї–µ–і—Б—В–≤–Є–µ –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –і–µ—Д–µ–Ї—В–Њ–≤ –≤ –Ї–Њ–ї–ї–∞–≥–µ–љ–∞—Е, –љ–Њ —В–∞–Ї–ґ–µ –≤—Б–ї–µ–і—Б—В–≤–Є–µ –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –і–µ—Д–µ–Ї—В–Њ–≤, –≤–ї–Є—П—О—Й–Є—Е –љ–∞ –±–Є–Њ—Б–Є–љ—В–µ–Ј, –њ–Њ—Б—В—В—А–∞–љ—Б–ї—П—Ж–Є–Њ–љ–љ—Л–µ –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є, —Б–µ–Ї—А–µ—Ж–Є—О, —Б–∞–Љ–Њ—Б–±–Њ—А–Ї—Г –Є —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є–µ –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤. –Т—Б–µ —Н—В–Є –њ—А–Њ—Ж–µ—Б—Б—Л, —А–∞—Б—Б–Љ–Њ—В—А–µ–љ–љ—Л–µ –љ–Є–ґ–µ, –њ–Њ–і–і–µ—А–ґ–Є–≤–∞—О—В—Б—П –Љ–љ–Њ–≥–Є–Љ–Є –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л–Љ–Є –±–µ–ї–Ї–∞–Љ–Є –Є –≥–µ–љ–∞–Љ–Є.
–Ч–∞ —Б–Є–љ—В–µ–Ј–Њ–Љ –њ—А–Њ–Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤ –љ–∞ —А–Є–±–Њ—Б–Њ–Љ–µ —Б–ї–µ–і—Г–µ—В –≥–Є–і—А–Њ–Ї—Б–Є–ї–Є—А–Њ–≤–∞–љ–Є–µ —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ–ї–Є–љ–Њ–≤ –Є –ї–Є–Ј–Є–љ–Њ–≤. –≠—В–Є –њ–Њ—Б—В—В—А–∞–љ—Б–ї—П—Ж–Є–Њ–љ–љ—Л–µ –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є–Є –Ј–∞–≤–Є—Б—П—В –Њ—В –∞—Б–Ї–Њ—А–±–Є–љ–Њ–≤–Њ–є –Ї–Є—Б–ї–Њ—В—Л –Є –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л –і–ї—П –њ—А–∞–≤–Є–ї—М–љ–Њ–є —Б–±–Њ—А–Ї–Є –њ–Њ–ї–Є–њ–µ–њ—В–Є–і–љ—Л—Е —Ж–µ–њ–µ–є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ –≤ –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–µ —Д–Є–±—А–Є–ї—Л. –У–Є–і—А–Њ–Ї—Б–Є–ї–Є—А–Њ–≤–∞–љ–Є–µ –њ—А–Њ–ї–Є–љ–Њ–≤ –Ї–∞—В–∞–ї–Є–Ј–Є—А—Г–µ—В—Б—П –њ—А–Њ–ї–Є–ї–3––≥–Є–і—А–Њ–Ї—Б–Є–ї–∞–Ј–Њ–є 1 (–≥–µ–љ LEPRE1/P3H1) –Є –Љ—Г—В–∞—Ж–Є–Є –≤ —Н—В–Њ–Љ –≥–µ–љ–µ –≤—Л–Ј—Л–≤–∞—О—В —А–µ—Ж–µ—Б—Б–Є–≤–љ–Њ–µ –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Њ–µ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–Њ –Ї–Њ—Б—В–µ–є [20]. –Ъ–Њ–ї–ї–∞–≥–µ–љ—Л —В–∞–Ї–ґ–µ —Б–Њ–і–µ—А–ґ–∞—В –≥–Є–і—А–Њ–Ї—Б–Є–ї–Є—А–Њ–≤–∞–љ–љ—Л–µ –ї–Є–Ј–Є–љ—Л, –Ї –Ї–Њ—В–Њ—А—Л–Љ –Ї–Њ–≤–∞–ї–µ–љ—В–љ–Њ –њ—А–Є–Ї—А–µ–њ–ї—П—О—В—Б—П –њ–Њ–ї–Є—Б–∞—Е–∞—А–Є–і—Л, —Б—В–∞–±–Є–ї–Є–Ј–Є—А—Г—О—Й–Є–µ –Є—Е —Б—В—А—Г–Ї—В—Г—А—Г. –У–Є–і—А–Њ–Ї—Б–Є–ї–Є—А–Њ–≤–∞–љ–Є–µ –њ–Њ–і–і–µ—А–ґ–Є–≤–∞–µ—В—Б—П –ї–Є–Ј–Є–љ–≥–Є–і—А–Њ–Ї—Б–Є–ї–∞–Ј–∞–Љ–Є (–≥–µ–љ—Л PLOD1, PLOD2, PLOD3), –Є –Љ—Г—В–∞—Ж–Є–Є –≤ —Н—В–Є—Е –≥–µ–љ–∞—Е –њ—А–Є–≤–Њ–і—П—В –Ї –і–Є—Б–њ–ї–∞–Ј–Є–Є –≠–ї–µ—А—Б–∞––Ф–∞–љ–ї–Њ [21] –Є –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Њ–Љ—Г —А–∞—Б—Б—В—А–Њ–є—Б—В–≤—Г –Ї–Њ—Б—В–µ–є [22]. –Ь–µ—В–∞–ї–ї–Њ–њ—А–Њ—В–µ–Є–љ–∞–Ј–∞ ADAMTS2 –≤—Л—А–µ–Ј–∞–µ—В N––њ—А–Њ–њ–µ–њ—В–Є–і —Г –њ—А–Њ–Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤ —В–Є–њ–Њ–≤ I, II, V, –Є –Љ—Г—В–∞—Ж–Є–Є –≤ —Н—В–Њ–Љ –≥–µ–љ–µ —В–∞–Ї–ґ–µ –≤—Л–Ј—Л–≤–∞—О—В –±–Њ–ї–µ–Ј–љ—М –≠–ї–µ—А—Б–∞––Ф–∞–љ–ї–Њ [23].
–Я–Њ–ї–Є–њ–µ–њ—В–Є–і–љ—Л–µ —Ж–µ–њ–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ —Б–∞–Љ–Њ—Б–Њ–±–Є—А–∞—О—В—Б—П –≤ –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–µ —Д–Є–±—А–Є–ї—Л, –Ї–Њ—В–Њ—А—Л–µ –≤–њ–Њ—Б–ї–µ–і—Б—В–≤–Є–Є –∞—Б—Б–Њ—Ж–Є–Є—А—Г—О—В—Б—П –≤ –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞. –Ы–Є–Ј–Є–ї–Њ–Ї—Б–Є–і–∞–Ј–∞ (–≥–µ–љ LOX), –∞ —В–∞–Ї–ґ–µ –ї–Є–Ј–Є–ї–Њ–Ї—Б–Є–і–∞–Ј–Њ–њ–Њ–і–Њ–±–љ—Л–µ —Д–µ—А–Љ–µ–љ—В—Л (–≥–µ–љ—Л LOXL1, LOXL2, LOXL3 –Є LOXL4) –Њ—Б—Г—Й–µ—Б—В–≤–ї—П—О—В –њ–Њ–њ–µ—А–µ—З–љ—Г—О —Б—И–Є–≤–Ї—Г –њ–Њ–ї–Є–њ–µ–њ—В–Є–і–љ—Л—Е —Ж–µ–њ–µ–є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞, —В–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ —Г—Б–Є–ї–Є–≤–∞—П –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї—Г—О –њ—А–Њ—З–љ–Њ—Б—В—М —Д–Є–±—А–Є–ї. –Ф–µ—Д–Є—Ж–Є—В –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є LOX –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–ї—Б—П —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б —Б–Є–љ–і—А–Њ–Љ–Њ–Љ –≠–ї–µ—А—Б–∞––Ф–∞–љ–ї–Њ [24]. –Ъ–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞ –њ—А–Є—Б–Њ–µ–і–Є–љ—П—В—Б—П –Ї –Ї–ї–µ—В–Њ—З–љ—Л–Љ –Љ–µ–Љ–±—А–∞–љ–∞–Љ —З–µ—А–µ–Ј –±–µ–ї–Ї–Є––∞–і–∞–њ—В–µ—А—Л, —В–∞–Ї–Є–µ –Ї–∞–Ї —Д–Є–±—А–Њ–љ–µ–Ї—В–Є–љ (–≥–µ–љ FN1) –Є –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–µ –Є–љ—В–µ–≥—А–Є–љ—Л (–±–Њ–ї–µ–µ 20 –≥–µ–љ–Њ–≤). –Я—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ –ї—О–Љ–Є–Ї–∞–љ (–≥–µ–љ LUM) –Є –±–µ–ї–Њ–Ї —Д–Є–±—А–Њ–Љ–Њ–і—Г–ї–Є–љ (FMOD) –≤–ї–Є—П—О—В –љ–∞ —Б–∞–Љ–Њ—Б–±–Њ—А–Ї—Г —Ж–µ–њ–µ–є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞, –Њ–≥—А–∞–љ–Є—З–Є–≤–∞—П —А–∞–Ј–Љ–µ—А —Д–Є–±—А–Є–ї. –≠–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В—Л –љ–∞ –ґ–Є–≤–Њ—В–љ—Л—Е —Г–Ї–∞–Ј—Л–≤–∞—О—В, —З—В–Њ –і–µ–ї–µ—Ж–Є–Є —Н—В–Є—Е –≥–µ–љ–Њ–≤ –њ—А–Є–≤–Њ–і—П—В –Ї —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ, –љ–∞–њ–Њ–Љ–Є–љ–∞—О—Й–Є–Љ –±–Њ–ї–µ–Ј–љ—М –≠–ї–µ—А—Б–∞––Ф–∞–љ–ї–Њ –Є –і—А—Г–≥–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є —З–µ–ї–Њ–≤–µ–Ї–∞ [17].
–†–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є–µ (—В–Њ –µ—Б—В—М –і–µ–≥—А–∞–і–∞—Ж–Є—П –Є–ї–Є –њ—А–Њ—В–µ–Њ–ї–Є–Ј) –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ –Т–Ъ–Ь –њ—А–Њ–Є–Ј–≤–Њ–і–Є—В—Б—П –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ –Љ–∞—В—А–Є–Ї—Б–љ—Л—Е –Љ–µ—В–∞–ї–ї–Њ–њ—А–Њ—В–µ–Є–љ–∞–Ј (–Ь–Ь–Я). –Р–Ї—В–Є–≤–љ–Њ—Б—В—М —А–∞–Ј–ї–Є—З–љ—Л—Е –Ь–Ь–Я –Є–Љ–µ–µ—В —З—А–µ–Ј–≤—Л—З–∞–є–љ–Њ —И–Є—А–Њ–Ї–Є–є —Б–њ–µ–Ї—В—А –±–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –њ–Њ—Б–ї–µ–і—Б—В–≤–Є–є, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Њ–љ–Є –і–µ–≥—А–∞–і–Є—А—Г—О—В –±–Њ–ї—М—И–Є–љ—Б—В–≤–Њ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж—Л: –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–µ –Ї–Њ–ї–ї–∞–≥–µ–љ—Л –Є –Ї–Њ–ї–ї–∞–≥–µ–љ—Л –±–∞–Ј–∞–ї—М–љ–Њ–є –Љ–µ–Љ–±—А–∞–љ—Л, –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ—Л, –і–µ–Ї–Њ—А–Є–љ, —Д–Є–±—А–Њ–Љ–Њ–і—Г–ї–Є–љ, —Д–Є–±—А–Њ–љ–µ–Ї—В–Є–љ –Є —В.–і. [25,26]. –Т –≥–µ–љ–Њ–Љ–µ —З–µ–ї–Њ–≤–µ–Ї–∞ –њ—А–Є—Б—Г—В—Б—В–≤—Г—О—В –љ–µ –Љ–µ–љ–µ–µ 200 –Ь–Ь–Я––њ–Њ–і–Њ–±–љ—Л—Е –≥–µ–љ–Њ–≤, –≤–Ї–ї—О—З–∞—П —Б–Њ–±—Б—В–≤–µ–љ–љ–Њ –Ь–Ь–Я (25 –≥–µ–љ–Њ–≤), –Љ–µ–Љ–±—А–∞–љ–љ–Њ–—Б–≤—П–Ј–∞–љ–љ—Л–µ –Ь–Ь–Я, ADAM –њ—А–Њ—В–µ–Є–љ–∞–Ј—Л (–і–Є–Ј–Є–љ—В–µ–≥—А–Є–љ––Љ–µ—В–∞–ї–ї–Њ–њ—А–Њ—В–µ–Є–љ–∞–Ј–љ—Л–µ –і–Њ–Љ–µ–љ—Л), ADAMTS –њ—А–Њ—В–µ–Є–љ–∞–Ј—Л (–і–Є–Ј–Є–љ—В–µ–≥—А–Є–љ––Љ–µ—В–∞–ї–ї–Њ–њ—А–Њ—В–µ–Є–љ–∞–Ј–љ—Л–µ –і–Њ–Љ–µ–љ—Л —Б —В—А–Њ–Љ–±–Њ—Б–њ–Њ–љ–і–Є–љ–Њ–≤—Л–Љ –Љ–Њ—В–Є–≤–Њ–Љ) –Є —А—П–і –і—А—Г–≥–Є—Е.
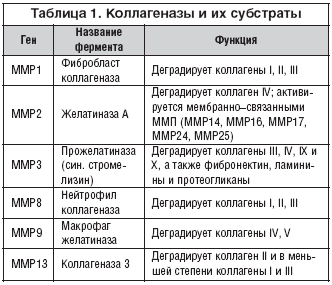
–Э–µ—Б–±–∞–ї–∞–љ—Б–Є—А–Њ–≤–∞–љ–љ—Л–є –њ—А–Њ—В–µ–Њ–ї–Є–Ј –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤ –Т–Ъ–Ь, –њ–Њ—А–Њ–ґ–і–∞–µ–Љ—Л–є –Є–Ј–±—Л—В–Њ—З–љ–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О –Ь–Ь–Я, –±—Л–ї —Б–≤—П–Ј–∞–љ —Б —А—П–і–Њ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є (–≤ —В–Њ–Љ —З–Є—Б–ї–µ, –∞—А—В—А–Є—В, —А–∞–Ї, –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј, –∞–љ–µ–≤—А–Є–Ј–Љ –∞–Њ—А—В—Л –Є —Д–Є–±—А–Њ–Ј) [27]. –Р–Ї—В–Є–≤–љ–Њ—Б—В—М –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ—Л—Е –Ь–Ь–Я –Љ–Њ–ґ–µ—В —А–µ–≥—Г–ї–Є—А–Њ–≤–∞—В—М—Б—П –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є—П–Љ–Є —Б–Њ —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–Љ–Є –Є–љ–≥–Є–±–Є—В–Њ—А–∞–Љ–Є —В–Ї–∞–љ–µ–≤—Л—Е –Љ–µ—В–∞–ї–ї–Њ–њ—А–Њ—В–µ–Є–љ–∞–Ј (TIMP –±–µ–ї–Ї–Є). –Ф–ї—П –Ї–∞–ґ–і–Њ–є —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–є –Ь–Ь–Я —Б—Г—Й–µ—Б—В–≤—Г–µ—В —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–є TIMP –±–µ–ї–Њ–Ї (–љ–∞–њ—А–Є–Љ–µ—А, –і–ї—П MMP1 –Є–љ–≥–Є–±–Є—В–Њ—А TIMP1 –Є —В.–і.). –°–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ –Ь–Ь–Я, –Ї–Њ—В–Њ—А—Л–µ –і–µ–≥—А–∞–і–Є—А—Г—О—В –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞, —В–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ —Г–і–∞–ї—П—П –Њ—Б–љ–Њ–≤–љ—Л–µ —Б—В—А—Г–Ї—В—Г—А–љ—Л–µ –Њ–њ–Њ—А—Л —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–µ–є —В–Ї–∞–љ–Є –Є–Ј–≤–µ—Б—В–љ—Л –њ–Њ–і –љ–∞–Ј–≤–∞–љ–Є–µ–Љ –Ї–Њ–ї–ї–∞–≥–µ–љa–Ј (—В–∞–±–ї. 1). –Т–∞–ґ–љ–Њ –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –≤—Б–µ –≤–љ–µ–Љ–µ–Љ–±—А–∞–љ–љ—Л–µ –Ь–Ь–Я —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П –≤–µ—Б—М–Љ–∞ —Б—Е–Њ–і–љ–Њ–є –њ–Њ–ї–љ–Њ–∞—В–Њ–Љ–љ–Њ–є —Б—В—А—Г–Ї—В—Г—А–Њ–є –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ—Л—Е –≥–ї–Њ–±—Г–ї, –Є –Ї–∞–ґ–і–∞—П –≥–ї–Њ–±—Г–ї–∞ —Д–µ—А–Љ–µ–љ—В–∞ –≤–Ї–ї—О—З–∞–µ—В —З–µ—В—Л—А–µ –Њ–±—П–Ј–∞—В–µ–ї—М–љ—Л—Е Ca2+ –Є –і–≤–∞ Zn2+ –Є–Њ–љ–∞ (—А–Є—Б. 3).
–≠–ї–∞—Б—В–Є–љ–Њ–≤—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞ –Т–Ъ–Ь –њ—А–Є–і–∞—О—В —Н–ї–∞—Б—В–Є—З–љ–Њ—Б—В—М –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж–µ –Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ю—Б–љ–Њ–≤–љ–Њ–є –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В —Н—В–Є—Е –≤–Њ–ї–Њ–Ї–Њ–љ – —Н–ї–∞—Б—В–Є–љ. –≠–ї–∞—Б—В–Є–љ —Б–Њ—Б—В–∞–≤–ї—П–µ—В –њ—А–Є–±–ї–Є–Ј–Є—В–µ–ї—М–љ–Њ 50% —Б—Г—Е–Њ–≥–Њ –≤–µ—Б–∞ –∞—А—В–µ—А–Є–є. –Т –Њ—В–ї–Є—З–Є–µ –Њ—В –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤ —Н–ї–∞—Б—В–Є–љ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ —В–Њ–ї—М–Ї–Њ –Њ–і–љ–Є–Љ –≥–µ–љ–Њ–Љ (ELN –љ–∞ —Е—А–Њ–Љ–Њ—Б–Њ–Љ–µ 7). –Ь—Г—В–∞—Ж–Є–Є –і–∞–љ–љ–Њ–≥–Њ –≥–µ–љ–∞ –њ—А–Є–≤–Њ–і—П—В –Ї —Б—В–µ–љ–Њ–Ј—Г –∞–Њ—А—В—Л [28] –Є –і—А—Г–≥–Є—Е –∞—А—В–µ—А–Є–є –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —З—А–µ–Ј–Љ–µ—А–љ–Њ–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –Ї–ї–µ—В–Њ–Ї –≥–ї–∞–і–Ї–Њ–є –Љ—Л—И–µ—З–љ–Њ–є —В–Ї–∞–љ–Є –≤ —Б—В–µ–љ–Ї–∞—Е –∞—А—В–µ—А–Є–є.
–Т –Њ—В–ї–Є—З–Є–µ –Њ—В –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤ –њ–Њ–ї–Є–њ–µ–њ—В–Є–і–љ—Л–µ —Ж–µ–њ–Є —Н–ї–∞—Б—В–Є–љ–∞ –љ–µ –≥–ї–Є–Ї–Њ–Ј–Є–ї–Є—А—Г—О—В—Б—П –Є –љ–µ —Б–Њ–і–µ—А–ґ–∞—В –≥–Є–і—А–Њ–Ї—Б–Є–ї–Є–Ј–Є–љ–Њ–≤. –Я–Њ—Б–ї–µ —Б–Є–љ—В–µ–Ј–∞ –љ–∞ —А–Є–±–Њ—Б–Њ–Љ–µ –Є –≥–Є–і—А–Њ–Ї—Б–Є–ї–Є—А–Њ–≤–∞–љ–Є—П –њ—А–Њ–ї–Є–љ–Њ–≤—Л—Е –Њ—Б—В–∞—В–Ї–Њ–≤ –њ—А–µ–Ї—Г—А—Б–Њ—А —Н–ї–∞—Б—В–Є–љ–∞ —Б–µ–Ї—А–µ—В–Є—А—Г–µ—В—Б—П –≤ –Т–Ъ–Ь, –≥–і–µ –Њ–љ –њ—А–Њ—Е–Њ–і–Є—В —З–µ—А–µ–Ј —Б–∞–Љ–Њ—Б–±–Њ—А–Ї—Г –Є –њ–Њ–њ–µ—А–µ—З–љ—Г—О —Б—И–Є–≤–Ї—Г –Љ–µ–ґ–і—Г –ї–Є–Ј–Є–љ–Њ–≤—Л–Љ–Є –Њ—Б—В–∞—В–Ї–∞–Љ–Є [14]. –У–Є–і—А–Њ–Ї—Б–Є–ї–Є—А–Њ–≤–∞–љ–Є–µ –њ—А–Њ–ї–Є–љ–Њ–≤ –Є –њ–Њ–њ–µ—А–µ—З–љ–∞—П —Б—И–Є–≤–Ї–∞ –≤—Л–њ–Њ–ї–љ—П—О—В—Б—П —З–µ—А–µ–Ј —В–µ –ґ–µ –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є–µ –њ—Г—В–Є, —З—В–Њ –Є –і–ї—П –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤.
–У–Є–±–Ї–Є–µ —Н–ї–∞—Б—В–Є–љ–Њ–≤—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞ –љ–µ —Б—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ—Л –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –ї–Є—И—М —Н–ї–∞—Б—В–Є–љ–Њ–Љ. –°–Њ—Б—В–Њ—П—Й–∞—П –Є–Ј —Н–ї–∞—Б—В–Є–љ–Њ–≤—Л—Е —Ж–µ–њ–µ–є —Б–µ—А–і—Ж–µ–≤–Є–љ–∞ —Н–ї–∞—Б—В–Є–љ–Њ–≤—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ –Ј–∞—Й–Є—Й–µ–љ–∞ —Б–љ–∞—А—Г–ґ–Є –≥–ї–Є–Ї–Њ–±–µ–ї–Ї–∞–Љ–Є –Љ–Є–Ї—А–Њ—Д–Є–±—А–Є–ї, –Ї–Њ—В–Њ—А—Л–µ –≤–Ї–ї—О—З–∞—О—В —Д–Є–±—А–Є–ї–ї–Є–љ—Л (–≥–µ–љ—Л FBN1, FBN3), —Д–Є–±—Г–ї–Є–љ—Л (–≥–µ–љ—Л FBLN1, FBLN2, FBLN5) –Є —Н–Љ–Є–ї–Є–љ—Л (EMILIN1, EMILIN2, EMILIN3, EMILIN4). –≠—В–Є —Б—А–∞–≤–љ–Є—В–µ–ї—М–љ–Њ –Љ–∞–ї–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–љ—Л–µ –±–µ–ї–Ї–Є —А–µ–≥—Г–ї–Є—А—Г—О—В –Є–љ—В–µ—А—Д–µ–є—Б –Љ–µ–ґ–і—Г —Н–ї–∞—Б—В–Є–љ–Њ–≤–Њ–є —Б–µ—А–і—Ж–µ–≤–Є–љ–Њ–є –Є –Љ–Є–Ї—А–Њ—Д–Є–±—А–Є–ї–ї–∞–Љ–Є, –∞ —В–∞–Ї–ґ–µ –њ–Њ–Ј–≤–Њ–ї—П—О—В –Њ—Б—Г—Й–µ—Б—В–≤–ї—П—В—М —В–Њ–љ–Ї—Г—О –њ–Њ–і—Б—В—А–Њ–є–Ї—Г —Н–ї–∞—Б—В–Є—З–љ–Њ—Б—В–Є –≤–Њ–ї–Њ–Ї–Њ–љ. –Ь—Л—И–Є —Б –і–µ–ї–µ—Ж–Є–µ–є –≥–µ–љ–∞ EMILIN1 –Є–Љ–µ—О—В –њ–Њ–≤—Л—И–µ–љ–љ–Њ–µ –Ї—А–Њ–≤—П–љ–Њ–µ –і–∞–≤–ї–µ–љ–Є–µ –≤—Б–ї–µ–і—Б—В–≤–Є–µ –≤–Њ–Ј—А–∞—Б—В–∞—О—Й–µ–≥–Њ —Б–Њ–њ—А–Њ—В–Є–≤–ї–µ–љ–Є—П –≤ –њ–µ—А–Є—Д–µ—А–Є–є–љ–Њ–є —Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–Є—Б—В–µ–Љ–µ –Є —Б—Г–ґ–µ–љ–љ–Њ–≥–Њ –њ—А–Њ—Б–≤–µ—В–∞ —Б–Њ—Б—Г–і–Њ–≤ [29].
–У–Є–±–Ї–Є–µ –≤–Њ–ї–Њ–Ї–љ–∞ —А–µ–Љ–Њ–і–µ–ї–Є—А—Г—О—В—Б—П (–і–µ–≥—А–∞–і–Є—А—Г—О—В—Б—П) —Н–ї–∞—Б—В–∞–Ј–∞–Љ–Є (–≥–µ–љ—Л ELA2A, ELA2B ELA3B, ELA3A, ELA1, ELA2). –§–∞–Ї—В–Є—З–µ—Б–Ї–Є –љ–µ–Ї–Њ—В–Њ—А—Л–µ –Є–Ј –Ь–Ь–Я – —В–∞–Ї–ґ–µ —Н–ї–∞—Б—В–∞–Ј—Л (–љ–∞–њ—А–Є–Љ–µ—А, —Д–µ—А–Љ–µ–љ—В MMP12 –Є–Ј–≤–µ—Б—В–µ–љ, –Ї–∞–Ї «—Н–ї–∞—Б—В–∞–Ј–∞ –Љ–∞–Ї—А–Њ–±–∞–Ї—В–µ—А–Є–Њ—Д–∞–≥–Њ–≤»).
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ–Њ–µ –≤—Л—И–µ –Ї—А–∞—В–Ї–Њ–µ –Њ–њ–Є—Б–∞–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–є –±–µ–ї–Ї–Њ–≤ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж—Л –њ–Њ–Ј–≤–Њ–ї—П–µ—В —Б—Д–Њ—А–Љ—Г–ї–Є—А–Њ–≤–∞—В—М –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л, —З–µ—А–µ–Ј –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–≥–ї–∞ –±—Л –≤–Њ–Ј–љ–Є–Ї–љ—Г—В—М –і–Є—Б–њ–ї–∞–Ј–Є—П —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ы—О–±–Њ–є –і–Є—Б–±–∞–ї–∞–љ—Б –≤ —Н—В–Њ–є —В–Њ–љ–Ї–Њ –љ–∞—Б—В—А–Њ–µ–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ–µ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є, –±—Г–і—М —В–Њ –∞–љ–Њ–Љ–∞–ї—М–љ–∞—П –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є—П —В–Ї–∞–љ–Є (–≤—Б–ї–µ–і—Б—В–≤–Є–µ, –љ–∞–њ—А–Є–Љ–µ—А, –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –і–µ—Д–µ–Ї—В–Њ–≤ –≤ –≥–µ–љ–µ TGFBR1 —А–µ—Ж–µ–њ—В–Њ—А–∞ TGF–? [30]), –Є–Ј–±—Л—В–Њ—З–љ–∞—П –і–µ–≥—А–∞–і–∞—Ж–Є—П –Ї–Њ–ї–ї–∞–≥–µ–љo–≤, –і–µ—Д–µ–Ї—В—Л –≤ —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –≥–µ–љ–∞—Е (–њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ–∞—Е, –Ї–Њ–ї–ї–∞–≥–µ–љ–∞—Е, —Н–ї–∞—Б—В–Є–љ–∞—Е –Є —В.–і.) –Є–ї–Є –∞–љ–Њ–Љ–∞–ї–Є–Є –≤ –њ–Њ—Б—В—В—А–∞–љ—Б–ї—П—Ж–Є–Њ–љ–љ—Л—Е –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є—П—Е, –Љ–Њ–ґ–µ—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї –Ф–°–Ґ. –Ф–ї—П —В–Њ–≥–Њ, —З—В–Њ–±—Л –Њ–≥—А–∞–љ–Є—З–Є—В—М—Б—П –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞–Љ–Є, –Ї–Њ—В–Њ—А—Л–µ —Б–≤—П–Ј—Л–≤–∞—О—В –Є–Љ–µ–љ–љ–Њ –і–µ—Д–Є—Ж–Є—В –Љ–∞–≥–љ–Є—П –Є —Н—В–Є–Њ–ї–Њ–≥–Є—О –Ф–°–Ґ, –Љ—Л –њ—А–Њ–∞–љ–∞–ї–Є–Ј–Є—А—Г–µ–Љ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ –Љ–∞–≥–љ–Є—П, –∞ –Ј–∞—В–µ–Љ –њ—А–µ–і—Б—В–∞–≤–Є–Љ —Б–Є–љ—В–µ–Ј –Є–Љ–µ—О—Й–Є—Е—Б—П –і–∞–љ–љ—Л—Е.
–Ь–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л
–≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ –Љ–∞–≥–љ–Є—П
–°—А–µ–і–Є –Ї–∞—В–Є–Њ–љ–Њ–≤, –њ—А–Є—Б—Г—В—Б—В–≤—Г—О—Й–Є—Е –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ —З–µ–ї–Њ–≤–µ–Ї–∞, –Є–Њ–љ –Љ–∞–≥–љ–Є—П (Mg2+) –љ–∞—Е–Њ–і–Є—В—Б—П –љ–∞ —З–µ—В–≤–µ—А—В–Њ–Љ –Љ–µ—Б—В–µ –њ–Њ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В–Є (–њ–Њ—Б–ї–µ –љ–∞—В—А–Є—П, –Ї–∞–ї–Є—П –Є –Ї–∞–ї—М—Ж–Є—П). Mg2+ —Б—Г—Й–µ—Б—В–≤–µ–љ–µ–љ –і–ї—П –∞–і–≥–µ–Ј–Є–Є –Є –Љ–Є–≥—А–∞—Ж–Є–Є –Ї–ї–µ—В–Њ–Ї, —Н–љ–µ—А–≥–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞, —В—А–∞–љ—Б–Ї—А–Є–њ—Ж–Є–Є –Ф–Э–Ъ, —Б—В–∞–±–Є–ї—М–љ–Њ—Б—В–Є –†–Э–Ъ, –±–µ–ї–Ї–Њ–≤–Њ–≥–Њ —Б–Є–љ—В–µ–Ј–∞, –∞ —В–∞–Ї–ґ–µ –і—А—Г–≥–Є—Е –Ї–ї–µ—В–Њ—З–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є [31–33].
–Т –∞–љ–љ–Њ—В–Є—А–Њ–≤–∞–љ–љ–Њ–є –њ–Њ—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—М–љ–Њ—Б—В–Є –≥–µ–љ–Њ–Љ–∞ —З–µ–ї–Њ–≤–µ–Ї–∞ —Б—Г—Й–µ—Б—В–≤—Г–µ—В –љ–µ –Љ–µ–љ–µ–µ 290 –≥–µ–љ–Њ–≤ –Є –±–µ–ї–Ї–Њ–≤—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤, –Њ –Ї–Њ—В–Њ—А—Л—Е –Є–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –Њ–љ–Є —Б–≤—П–Ј—Л–≤–∞—О—В Mg2+ –Ї–∞–Ї –Ї–Њ—Д–∞–Ї—В–Њ—А. –Ґ–∞–Ї –Ї–∞–Ї –≤ –≥–µ–љ–Њ–Љ–µ —З–µ–ї–Њ–≤–µ–Ї–∞ –Є–Ј–≤–µ—Б—В–љ–∞ —Д—Г–љ–Ї—Ж–Є—П –Љ–µ–љ–µ–µ —З–µ–Љ –њ–Њ–ї–Њ–≤–Є–љ—Л –≤—Б–µ—Е –≥–µ–љ–Њ–≤ (14000 –Є–Ј 31000), –Љ–Њ–ґ–љ–Њ –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В—М, —З—В–Њ —З–Є—Б–ї–Њ –Љ–∞–≥–љ–Є–є––Ј–∞–≤–Є—Б–Є–Љ—Л—Е –±–µ–ї–Ї–Њ–≤ –±—Г–і–µ—В –њ—А–µ–≤—Л—И–∞—В—М 500. –Ґ–∞–Ї —З—В–Њ –љ–∞ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ–Љ —Г—А–Њ–≤–љ–µ —Б–њ–µ–Ї—В—А –≤–ї–Є—П–љ–Є—П –Љ–∞–≥–љ–Є—П –љ–∞ —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—О —З–µ–ї–Њ–≤–µ–Ї–∞ –µ–і–≤–∞ –ї–Є –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ–њ–Є—Б–∞–љ –≤ –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е —Б–ї–Њ–≤–∞—Е.
–° —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є —В–Њ—З–Ї–Є –Ј—А–µ–љ–Є—П –і–Њ 53% –Љ–∞–≥–љ–Є—П –Ї–Њ–љ—Ж–µ–љ—В—А–Є—А—Г–µ—В—Б—П –≤ –Ї–Њ—Б—В–љ–Њ–є —В–Ї–∞–љ–Є, –і–µ–љ—В–Є–љ–µ –Є —Н–Љ–∞–ї–Є –Ј—Г–±–Њ–≤ –Є –Њ–Ї–Њ–ї–Њ 20% – –≤ —В–Ї–∞–љ—П—Е —Б –≤—Л—Б–Њ–Ї–Њ–є –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О (–Љ–Њ–Ј–≥, —Б–µ—А–і—Ж–µ, –Љ—Л—И—Ж—Л, –љ–∞–і–њ–Њ—З–µ—З–љ–Є–Ї–Є, –њ–Њ—З–Ї–Є, –њ–µ—З–µ–љ—М). –Ґ–Њ–ї—М–Ї–Њ 10% –≤—Б–µ–≥–Њ –Љ–∞–≥–љ–Є—П –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ —З–µ–ї–Њ–≤–µ–Ї–∞ –љ–∞—Е–Њ–і–Є—В—Б—П –≤–љ–µ –Ї–ї–µ—В–Њ–Ї, –Є 90% –Љ–∞–≥–љ–Є–µ–≤—Л—Е –Є–Њ–љ–Њ–≤ –Ї–Њ–љ—Ж–µ–љ—В—А–Є—А—Г—О—В—Б—П –≤–љ—Г—В—А–Є –Ї–ї–µ—В–Њ–Ї –≤ —Д–Њ—А–Љ–µ Mg2+•AT–§ (30% –≤ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є—П—Е, 50% –≤ —Ж–Є—В–Њ–Ј–Њ–ї–µ –Є 10% –≤ —П–і—А–µ). –£ –Ј–і–Њ—А–Њ–≤–Њ–≥–Њ —З–µ–ї–Њ–≤–µ–Ї–∞ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –Љ–∞–≥–љ–Є—П –≤ —Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –њ–Њ–і–і–µ—А–ґ–Є–≤–∞–µ—В—Б—П –≤ –і–Њ—Б—В–∞—В–Њ—З–љ–Њ —Г–Ј–Ї–Њ–Љ –і–Є–∞–њ–∞–Ј–Њ–љ–µ (–љ–Њ—А–Љ–∞ 0,7–1,1 –Љ–Љ–Њ–ї—М/–ї). –≠—В–Њ—В –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ—Л–є –Љ–∞–≥–љ–Є–є –љ–∞—Е–Њ–і–Є—В—Б—П –≤ –љ–µ–њ—А–µ—А—Л–≤–љ–Њ–Љ –Њ–±–Љ–µ–љ–µ —Б –Љ–∞–≥–љ–Є–µ–≤—Л–Љ–Є –Ј–∞–њ–∞—Б–∞–Љ–Є –Ї–Њ—Б—В–µ–є –Є –Љ—Л—И–µ—З–љ–Њ–є —В–Ї–∞–љ–Є. –°–±–∞–ї–∞–љ—Б–Є—А–Њ–≤–∞–љ–љ–∞—П –і–Є–µ—В–∞ –і–Њ–ї–ґ–љ–∞ —Б–Њ–і–µ—А–ґ–∞—В—М ?400 –Љ–≥ –Љ–∞–≥–љ–Є—П –≤ —Б—Г—В–Ї–Є, –Є–Ј –Ї–Њ—В–Њ—А–Њ–≥–Њ –∞–і—Б–Њ—А–±–Є—А—Г–µ—В—Б—П, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –Њ–Ї–Њ–ї–Њ 200 –Љ–≥.
–£–Љ–µ–љ—М—И–µ–љ–Є–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –µ–ґ–µ–і–љ–µ–≤–љ–Њ –њ—А–Є–љ–Є–Љ–∞–µ–Љ–Њ–≥–Њ –Љ–∞–≥–љ–Є—П –Љ–Њ–ґ–µ—В –Ї–Њ–Љ–њ–µ–љ—Б–Є—А–Њ–≤–∞—В—М—Б—П –≤–Њ–Ј—А–∞—Б—В–∞—О—Й–µ–є –∞–і—Б–Њ—А–±—Ж–Є–µ–є –Љ–∞–≥–љ–Є—П –≤ –Ї–Є—И–µ—З–љ–Є–Ї–µ –Є —Г–Љ–µ–љ—М—И–µ–љ–Є–µ–Љ –≤—Л–і–µ–ї–µ–љ–Є—П –µ–≥–Њ —З–µ—А–µ–Ј –њ–Њ—З–Ї–Є. –≠—В–Є –њ—А–Њ—Ж–µ—Б—Б—Л —В—А–∞–љ—Б–њ–Њ—А—В–∞ Mg2+ —А–µ–≥—Г–ї–Є—А—Г—О—В—Б—П —А—П–і–Њ–Љ –≥–Њ—А–Љ–Њ–љ–Њ–≤, –≤–Ї–ї—О—З–∞—П –∞–љ—В–Є–і–Є—Г—А–µ—В–Є—З–µ—Б–Ї–Є–є –њ–µ–њ—В–Є–і, –≥–ї—О–Ї–∞–≥–Њ–љ, –Ї–∞–ї—М—Ж–Є—В–Њ–љ–Є–љ, –≥–Њ—А–Љ–Њ–љ –њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ–Њ–є –ґ–µ–ї–µ–Ј—Л (–њ–∞—А–∞—В–≥–Њ—А–Љ–Њ–љ) –Є –Є–љ—Б—Г–ї–Є–љ [34]. –Ф–µ—Д–Є—Ж–Є—В –Љ–∞–≥–љ–Є—П –Љ–Њ–ґ–µ—В —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—В—М—Б—П –≤—В–Њ—А–Є—З–љ—Л–Љ–Є –Є–Њ–љ––і–µ—Д–Є—Ж–Є—В–∞–Љ–Є, –≤–Ї–ї—О—З–∞—П –≥–Є–њ–Њ–Ї–∞–ї–Є–µ–Љ–Є—О, –≥–Є–њ–Њ—Д–Њ—Б—Д–∞—В–µ–Љ–Є—О –Є –≥–Є–њ–Њ–Ї–∞–ї—М—Ж–Є–µ–Љ–Є—О. –•—А–Њ–љ–Є—З–µ—Б–Ї–Є–є –і–µ—Д–Є—Ж–Є—В –Љ–∞–≥–љ–Є—П –Љ–Њ–ґ–µ—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї –∞–љ–Њ—А–µ–Ї—Б–Є–Є, —В–Њ—И–љ–Њ—В–µ –Є –њ–µ—А–Є–Њ–і–Є—З–µ—Б–Ї–Њ–є —Б–ї–∞–±–Њ—Б—В–Є, –Ї –Њ–±—Й–µ–Љ—Г —Б–љ–Є–ґ–µ–љ–Є—О —В–Њ–љ—Г—Б–∞ –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А—Л, —В–∞—Е–Є–Ї–∞—А–і–Є–Є, —Б—Г–і–Њ—А–Њ–≥–∞–Љ –≤ –Љ—Л—И—Ж–∞—Е, —А–µ–Ј–Ї–Њ –≤—Л—А–∞–ґ–µ–љ–љ–Њ–є –∞—Б—В–µ–љ–Є–Ј–∞—Ж–Є–Є, –≤–њ–ї–Њ—В—М –і–Њ —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—П —Б–Є–љ–і—А–Њ–Љ–∞ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є —Г—Б—В–∞–ї–Њ—Б—В–Є [35].
–§–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є –≥–Є–њ–Њ–Љ–∞–≥–љ–µ–Ј–Є—П –Љ–Њ–ґ–µ—В –±—Л—В—М —Б–ї–µ–і—Б—В–≤–Є–µ–Љ —Г–Љ–µ–љ—М—И–µ–љ–љ–Њ–≥–Њ –њ—А–Є–µ–Љ–∞ –њ–Є—Й–µ–≤–Њ–≥–Њ –Љ–∞–≥–љ–Є—П, –њ–ї–Њ—Е–Њ–є –∞–і—Б–Њ—А–±—Ж–Є–Є –≤ —В–Њ–љ–Ї–Њ–Љ –Ї–Є—И–µ—З–љ–Є–Ї–µ –Є–ї–Є —Г–≤–µ–ї–Є—З–µ–љ–Є—П –≤—Л–і–µ–ї–µ–љ–Є—П —З–µ—А–µ–Ј –њ–Њ—З–Ї–Є. –Ф–µ—Д–Є—Ж–Є—В –Љ–∞–≥–љ–Є—П –Љ–Њ–ґ–µ—В –±—Л—В—М –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–Љ –Є–ї–Є –±—Л—В—М —Б–≤—П–Ј–∞–љ–љ—Л–Љ —Б –≤–љ–µ—И–љ–Є–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є (–љ–µ—Б–±–∞–ї–∞–љ—Б–Є—А–Њ–≤–∞–љ–љ–Њ–µ –њ–Є—В–∞–љ–Є–µ, —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–є —Н–Љ–Њ—Ж–Є–Њ–љ–∞–ї—М–љ—Л–є —Б—В—А–µ—Б—Б). –Т –Њ–±—Й–Є—Е —З–µ—А—В–∞—Е, –Є–Ј–≤–µ—Б—В–љ—Л–µ –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –њ—А–Є–≤–Њ–і—П—Й–Є–µ –Ї –Њ—В—З–µ—В–ї–Є–≤–Њ–є –≥–Є–њ–Њ–Љ–∞–≥–љ–µ–Ј–Є–Є, —Б—А–∞–≤–љ–Є—В–µ–ї—М–љ–Њ —А–µ–і–Ї–Є (1:50000). –Ґ–µ–Љ –љ–µ –Љ–µ–љ–µ–µ –≥–Є–њ–Њ–Љ–∞–≥–љ–µ–Ј–Є—П —З–∞—Б—В–Њ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–µ—В—Б—П —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ 2 —В–Є–њ–∞, –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–µ–є, –Ш–С–°, –±—А–Њ–љ—Е–Є–∞–ї—М–љ–Њ–є –∞—Б—В–Љ–Њ–є. –Ю–±–Њ—Б—В—А–µ–љ–Є–µ –і–µ—Д–Є—Ж–Є—В–∞ –Љ–∞–≥–љ–Є—П —Б–Њ–≥–ї–∞—Б—Г–µ—В—Б—П —Б —Г—Е—Г–і—И–µ–љ–Є–µ–Љ –њ—А–Њ—В–µ–Ї–∞–љ–Є—П —Н—В–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –Є —А–∞–Ј–≤–Є—В–Є–µ–Љ –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є [35,36].
–С–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В—М –Љ–∞–≥–љ–Є—П –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ —А–µ–≥—Г–ї–Є—А—Г–µ—В—Б—П —А—П–і–Њ–Љ –≥–µ–љ–Њ–≤, —Б—А–µ–і–Є –Ї–Њ—В–Њ—А—Л—Е TRPM6 –Є TRPM7 –љ–∞–Є–±–Њ–ї–µ–µ –≤–∞–ґ–љ—Л. –С–µ–ї–Њ–Ї TRPM6 (–∞–љ–≥–ї. «transient receptor potential cation channel 6») —П–≤–ї—П–µ—В—Б—П –Є–Њ–љ–љ—Л–Љ –Ї–∞–љ–∞–ї–Њ–Љ, —В—А–∞–љ—Б–њ–Њ—А—В–Є—А—Г—О—Й–Є–Љ –і–≤—Г—Е–≤–∞–ї–µ–љ—В–љ—Л–µ –Ї–∞—В–Є–Њ–љ—Л. TRPM6 —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г–µ—В —Б –і—А—Г–≥–Є–Љ Mg2+––њ—А–Њ–љ–Є—Ж–∞–µ–Љ—Л–Љ –Ї–∞–љ–∞–ї–Њ–Љ TRPM7, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї —Б–±–Њ—А–Ї–µ —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е TRPM6/TRPM7 –Ї–Њ–Љ–њ–ї–µ–Ї—Б–Њ–≤ –љ–∞ –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–Є –Ї–ї–µ—В–Ї–Є [37]. –Ь—Г—В–∞—Ж–Є–Є –≤ TRPM6 –Љ–Њ–≥—Г—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї –≥–Є–њ–Њ–Љ–∞–≥–љ–µ–Ј–Є–Є –Є –≤—В–Њ—А–Є—З–љ–Њ–є –≥–Є–њ–Њ–Ї–∞–ї—М—Ж–Є–µ–Љ–Є–Є [38]. TRPM7 –Љ–Њ–ґ–µ—В –±—Л—В—М –≤–Њ–≤–ї–µ—З–µ–љ –≤ –і–µ—Д–Є—Ж–Є—В –Љ–∞–≥–љ–Є—П, —Б–≤—П–Ј–∞–љ–љ—Л–є —Б —Н–Љ–Њ—Ж–Є–Њ–љ–∞–ї—М–љ—Л–Љ —Б—В—А–µ—Б—Б–Њ–Љ –њ–Њ–і –і–µ–є—Б—В–≤–Є–µ–Љ –Ї–∞—В–µ—Е–Њ–ї–∞–Љ–Є–љ–Њ–≤ [39].
–Я—А–Є –≥–Є–њ–Њ–Љ–∞–≥–љ–µ–Ј–Є–Є —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—П –Є –і—А—Г–≥–Њ–≥–Њ –≥–µ–љ–∞ –Є–Њ–љ–љ–Њ–≥–Њ —В—А–∞–љ—Б–њ–Њ—А—В–µ—А–∞ SLC41A1 (—Б–Њ–ї—М–≤–∞—В–—В—А–∞–љ—Б–њ–Њ—А—В–µ—А —В–Є–њ–∞ 41.1): —Г –Љ—Л—И–µ–є –љ–∞ –±–µ–Ј–Љ–∞–≥–љ–Є–µ–≤–Њ–є –і–Є–µ—В–µ —Н–Ї—Б–њ—А–µ—Б—Б–Є—П –Љ–†–Э–Ъ –≥–µ–љ–∞ SLC41A1 —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –≤ –њ–Њ—З–Ї–∞—Е, –Ї–Є—И–µ—З–љ–Є–Ї–µ –Є —Б–µ—А–і—Ж–µ. –Т –і–Њ–њ–Њ–ї–љ–µ–љ–Є–µ –Ї Mg2+, SLC41A1 —В–∞–Ї–ґ–µ –Љ–Њ–ґ–µ—В —В—А–∞–љ—Б–њ–Њ—А—В–Є—А–Њ–≤–∞—В—М Sr2+, Zn2+, Cu2+ –Є Co2+ [40]. –Э–µ–±–Њ–ї—М—И–Є–µ –Љ–µ–Љ–±—А–∞–љ–љ—Л–µ –±–µ–ї–Ї–Є, –≤–Ї–ї—О—З–∞—О—Й–Є–µ –≤ —Б–µ–±—П FXYD––і–Њ–Љ–µ–љ—Л (–і–Њ–Љ–µ–љ—Л, —Б–Њ–і–µ—А–ґ–∞—Й–Є–µ —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–є —Д—А–∞–≥–Љ–µ–љ—В –∞–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В–љ–Њ–є –њ–Њ—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—М–љ–Њ—Б—В–Є– —Д–µ–љ–Є–ї–∞–ї–∞–љ–Є–љ–X–—В–Є—А–Њ–Ј–Є–љ––∞—Б–њ–∞—А—В–∞—В), —В–∞–Ї–ґ–µ –≤–Њ–≤–ї–µ—З–µ–љ—Л –≤ —В—А–∞–љ—Б–њ–Њ—А—В –Љ–∞–≥–љ–Є—П. –Ь—Г—В–∞—Ж–Є–Є –∞–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В–љ—Л—Е –Њ—Б—В–∞—В–Ї–Њ–≤ –≤ –±–µ–ї–Ї–µ FXYD2 –њ—А–Є–≤–Њ–і–Є–ї–Є –Ї –њ–Њ—З–µ—З–љ–Њ–є –≥–Є–њ–Њ–Љ–∞–≥–љ–µ–Ј–Є–Є [41,42].
–Ъ–ї–∞—Г–і–Є–љ—Л (–≥–µ–љ—Л CLDN16 –Є CLDN19) – —В—А–∞–љ—Б–Љ–µ–Љ–±—А–∞–љ–љ—Л–µ –±–µ–ї–Ї–Є, –Њ–±–љ–∞—А—Г–ґ–µ–љ–љ—Л–µ –љ–∞ –њ–ї–Њ—В–љ—Л—Е —Б–Њ–µ–і–Є–љ–µ–љ–Є—П—Е –Ї–ї–µ—В–Њ–Ї. –Я–ї–Њ—В–љ—Л–µ —Б–Њ–µ–і–Є–љ–µ–љ–Є—П –Ї–ї–µ—В–Њ–Ї (–∞–љ–≥–ї. «tight junction») –Њ–±—А–∞–Ј—Г—О—В –±–∞—А—М–µ—А—Л, –Ї–Њ—В–Њ—А—Л–µ –Ї–Њ–љ—В—А–Њ–ї–Є—А—Г—О—В —В—А–∞–љ—Б–њ–Њ—А—В –Є–Њ–љ–Њ–≤ –Є –Љ–µ—В–∞–±–Њ–ї–Є—В–Њ–≤ –њ–Њ–њ–µ—А–µ–Ї –ї–Є—Б—В–∞ —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї, –∞ —В–∞–Ї–ґ–µ –њ–µ—А–µ–Љ–µ—Й–µ–љ–Є–µ –±–µ–ї–Ї–Њ–≤ –Є –ї–Є–њ–Є–і–Њ–≤ –Љ–µ–ґ–і—Г –∞–њ–Є–Ї–∞–ї—М–љ—Л–Љ–Є –Є –±–∞–Ј–Њ–ї–∞—В–µ—А–∞–ї—М–љ—Л–Љ–Є –Њ–±–ї–∞—Б—В—П–Љ–Є —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї [43]. –У–µ–љ CLDN16 (–њ–∞—А–∞—Ж–µ–ї–ї–Є–љ 1) —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г–µ—В—Б—П –љ–∞ –њ–ї–Њ—В–љ—Л—Е —Б–Њ–µ–і–Є–љ–µ–љ–Є—П—Е –њ–Њ—З–µ—З–љ—Л—Е —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞ —В–Њ–ї—Б—В–Њ–Љ –≤–Њ—Б—Е–Њ–і—П—Й–µ–Љ –Њ—В—А–Њ—Б—В–Ї–µ –њ–µ—В–ї–Є –У–µ–љ–ї–µ, –Є –Є–≥—А–∞–µ—В —Ж–µ–љ—В—А–∞–ї—М–љ—Г—О —А–Њ–ї—М –≤ —А–µ–∞–±—Б–Њ—А–±—Ж–Є–Є –і–≤—Г—Е–≤–∞–ї–µ–љ—В–љ—Л—Е –Ї–∞—В–Є–Њ–љ–Њ–≤. –У–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –і–µ—Д–µ–Ї—В—Л –≤ –Ї–ї–∞—Г–і–Є–љ–µ–16 –±—Л–ї–Є —Б–≤—П–Ј–∞–љ—Л —Б –њ–µ—А–≤–Є—З–љ–Њ–є –≥–Є–њ–Њ–Љ–∞–≥–љ–µ–Ј–Є–µ–є [44], –∞ –і–µ—Д–µ–Ї—В—Л –Ї–ї–∞—Г–і–Є–љ–∞–19 –њ—А–Є–≤–Њ–і—П—В –Ї –њ–Њ—З–µ—З–љ–Њ–є –≥–Є–њ–Њ–Љ–∞–≥–љ–µ–Ј–Є–Є —Б –≤–Њ–≤–ї–µ—З–µ–љ–Є–µ–Љ –≥–ї–∞–Ј–љ–Њ–є —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–Є (–Љ–Є–Њ–њ–Є—П, –њ–Њ–і–≤—Л–≤–Є—Е —Е—А—Г—Б—В–∞–ї–Є–Ї–∞) [45].
–†–µ–≥—Г–ї–Є—А—Г–µ—В –Њ–±–Љ–µ–љ –Љ–∞–≥–љ–Є—П –Є Ca2+/Mg2+–—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ—Л–є —А–µ—Ж–µ–њ—В–Њ—А (–≥–µ–љ CASR) – G––±–µ–ї–Њ–Ї–—Б–≤—П–Ј–∞–љ–љ—Л–є —А–µ—Ж–µ–њ—В–Њ—А –њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Њ–є –Љ–µ–Љ–±—А–∞–љ—Л, –Ї–Њ—В–Њ—А—Л–є —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г–µ—В—Б—П –≤ –њ–∞—А–∞—Й–Є—В–Њ–≤–Є–і–љ–Њ–є –ґ–µ–ї–µ–Ј–µ –Є –≤ –њ–Њ—З–µ—З–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–∞—Е. –С–ї–∞–≥–Њ–і–∞—А—П –≤—Л—Б–Њ–Ї–Њ–є —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є –Ї –љ–µ–±–Њ–ї—М—И–Є–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ –≤ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П—Е —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—Й–Є—Е –Ї–∞–ї—М—Ж–Є—П –Є –Љ–∞–≥–љ–Є—П CASR –і–µ–є—Б—В–≤—Г–µ—В –Ї–∞–Ї —Б–µ–љ—Б–Њ—А (–і–∞—В—З–Є–Ї), —А–µ–∞–≥–Є—А—Г—О—Й–Є–є –љ–∞ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—О –Ї–∞—В–Є–Њ–љ–Њ–≤, –Є –Є–≥—А–∞–µ—В —Б—Г—Й–µ—Б—В–≤–µ–љ–љ—Г—О —А–Њ–ї—М –≤ –њ–Њ–і–і–µ—А–ґ–∞–љ–Є–Є –Ї–∞—В–Є–Њ–љ–љ–Њ–≥–Њ –≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ [46]. –Ф–µ—Д–µ–Ї—В—Л –≤ —Н—В–Њ–Љ –≥–µ–љ–µ —Б–≤—П–Ј–∞–љ—Л –Ї–∞–Ї —Б –≥–Є–њ–µ—А–Ї–∞–ї—М—Ж–Є–µ–Љ–Є–µ–є, —В–∞–Ї –Є —Б –≥–Є–њ–Њ–Ї–∞–ї—М—Ж–Є–µ–Љ–Є–µ–є [47]. –Р–Ї—В–Є–≤–∞—Ж–Є—П –≥–µ–љ–∞ CASR Ca2+/Mg2+–—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ–≥–Њ —А–µ—Ж–µ–њ—В–Њ—А–∞ —Г–Љ–µ–љ—М—И–∞–µ—В –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –±–µ–ї–Ї–Њ–≤–Њ–є –Ї–Є–љ–∞–Ј—Л –Р (PKA). –≠—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї —Г–Љ–µ–љ—М—И–µ–љ–Є—О —Д–Њ—Б—Д–Њ—А–Є–ї–Є—А–Њ–≤–∞–љ–Є—П –Ї–ї–∞—Г–і–Є–љ–∞–16, —В—А–∞–љ—Б–ї–Њ–Ї–∞—Ж–Є–Є –Ї–ї–∞—Г–і–Є–љ–∞–16 –≤ –ї–Є–Ј–Њ—Б–Њ–Љ—Л, –∞ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ – –Ї —Г–Љ–µ–љ—М—И–µ–љ–Є—О —А–µ–∞–±—Б–Њ—А–±—Ж–Є–Є –Љ–∞–≥–љ–Є—П –≤ –њ–Њ—З–µ—З–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–∞—Е [48].
–Ь–µ—В–∞–ї–ї–Њ—В–Є–Њ–љ–µ–љ–Є–љ 2A (MT2A), –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, –Є–≥—А–∞–µ—В –≤–∞–ґ–љ—Г—О —А–Њ–ї—М –≤ –Ј–∞—Й–Є—В–µ –Ї–ї–µ—В–Њ–Ї –Њ—В –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є [49]. –Т —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Г—Б–ї–Њ–≤–Є—П—Е –Ь–Ґ2–Р –≤–Њ–≤–ї–µ—З–µ–љ –≤ —А–µ–≥—Г–ї–Є—А–Њ–≤–∞–љ–Є–µ –≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ —Ж–Є–љ–Ї–∞ [50]. –Р–ї–ї–µ–ї—М G –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–∞ +838 C/G —Б–Њ–Њ—В–≤e—В—Б—В–≤—Г–µ—В –њ–Њ–≤—Л—И–µ–љ–Є—О –±–µ–ї–Ї–∞ –Њ—Б—В—А–Њ–є —Д–∞–Ј—Л MCP1 –Є —Г–Љ–µ–љ—М—И–µ–љ–Є—О —Г—А–Њ–≤–љ–µ–є —Ж–Є–љ–Ї–∞, –Љ–µ–і–Є –Є –Љ–∞–≥–љ–Є—П –≤ —Н—А–Є—В—А–Њ—Ж–Є—В–∞—Е, –≤–Њ–Ј—А–∞—Б—В–∞–љ–Є—О —Г—А–Њ–≤–љ—П –ґ–µ–ї–µ–Ј–∞ –≤ –њ–ї–∞–Ј–Љ–µ, –∞ —В–∞–Ї–ґ–µ –њ–Њ–≤—Л—И–µ–љ–љ–Њ–є —Б–Ї–ї–Њ–љ–љ–Њ—Б—В–Є –Ї –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—О –Љ—П–≥–Ї–Є—Е –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ—В–Є—З–µ—Б–Ї–Є—Е –±–ї—П—И–µ–Ї [51].
–Т–Њ–Ј–Љ–Њ–ґ–љ—Л–µ —А–Њ–ї–Є –Љ–∞–≥–љ–Є—П
–≤ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–µ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є
–Ґ–Њ—З–љ—Л–µ –Є —А–∞–Ј–љ–Њ—Б—В–Њ—А–Њ–љ–љ–Є–µ –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –і–∞–љ–љ—Л–µ –Њ –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –±—Л–ї–Є –±—Л –≤–µ—Б—М–Љ–∞ –њ–Њ–ї–µ–Ј–љ—Л –і–ї—П —А–∞—Б—Б–Љ–Њ—В—А–µ–љ–Є—П –љ–∞–Є–±–Њ–ї–µ–µ –≤–µ—А–Њ—П—В–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ –±–Њ–ї–µ–Ј–љ–Є. –£–≤—Л, —В—Й–∞—В–µ–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –≥–Є—Б—В–Њ–ї–Њ–≥–Є–Є –љ–Ф–°–Ґ –љ–µ –њ—А–Њ–≤–Њ–і–Є–ї–Њ—Б—М, —В–∞–Ї —З—В–Њ —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є, —Б–≤—П–Ј–∞–љ–љ—Л–µ –Є–Љ–µ–љ–љ–Њ —Б –љ–Ф–°–Ґ, –љ–µ–Є–Ј–≤–µ—Б—В–љ—Л. –Э–µ –Є–Љ–µ–µ—В—Б—П —В–∞–Ї–ґ–µ –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –њ—А–Є –і–µ—Д–Є—Ж–Є—В–µ –Љ–∞–≥–љ–Є—П, —Е–Њ—В—П –Є –Є–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –≥–Є–њ–Њ–Љ–∞–≥–љ–µ–Ј–Є—П –њ—А–Є–≤–Њ–і–Є—В –Ї –Є–Ј–Љ–µ–љ–µ–љ–Є—О –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Є—Е —Б–≤–Њ–є—Б—В–≤ –∞—А—В–µ—А–Є–є [52–54]. –Ь—Л –Љ–Њ–ґ–µ–Љ –і–Њ–њ—Г—Б—В–Є—В—М, —З—В–Њ –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ—А–Є –≥–Є–њ–Њ–Љ–∞–≥–љ–µ–Ј–Є—П—Е –≤—Б–µ–≥–і–∞ –±—Г–і—Г—В —Г—Е—Г–і—И–∞—В—М –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Є–µ —Б–≤–Њ–є—Б—В–≤–∞ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є (–њ—А–Њ—З–љ–Њ—Б—В—М, —Н–ї–∞—Б—В–Є—З–љ–Њ—Б—В—М) –Є –љ–∞ –Њ—Б–љ–Њ–≤–µ —Н—В–Њ–≥–Њ –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–Є—П —А–∞—Б—Б–Љ–Њ—В—А–µ—В—М —А–∞–Ј–ї–Є—З–љ—Л–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л–µ –њ—Г—В–Є, —З–µ—А–µ–Ј –Ї–Њ—В–Њ—А—Л–µ –і–µ—Д–Є—Ж–Є—В Mg2+ –Љ–Њ–ґ–µ—В –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ–Њ –њ–Њ–≤–ї–Є—П—В—М –љ–∞ —Б—В—А—Г–Ї—В—Г—А—Г —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є.
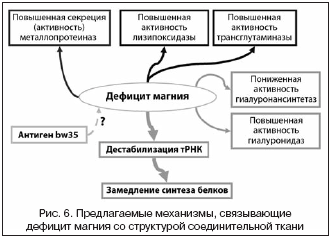
–Э–∞–Є–±–Њ–ї–µ–µ –Њ–±—Й–Є–є —Н—Д—Д–µ–Ї—В –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П Mg2+ –љ–∞ –ї—О–±—Г—О —В–Ї–∞–љ—М –Ј–∞–Ї–ї—О—З–∞–µ—В—Б—П –≤ —В–Њ–Љ, —З—В–Њ –Є–Њ–љ—Л Mg2+ –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л –і–ї—П —Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є –љ–µ–Ї–Њ–і–Є—А—Г—О—Й–Є—Е –†–Э–Ъ. –Т —З–∞—Б—В–љ–Њ—Б—В–Є, –Є–Њ–љ Mg2+ —Б—В–∞–±–Є–ї–Є–Ј–Є—А—Г—О—В —Б—В—А—Г–Ї—В—Г—А—Г —В—А–∞–љ—Б–њ–Њ—А—В–љ–Њ–є –†–Э–Ъ (—А–Є—Б. 4) –Є –і–µ—Д–Є—Ж–Є—В –Љ–∞–≥–љ–Є—П –њ—А–Є–≤–µ–і–µ—В –Ї —Г–≤–µ–ї–Є—З–µ–љ–Є—О —З–Є—Б–ї–∞ –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї —В–†–Э–Ъ, —В–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ —Б–љ–Є–ґ–∞—П –Є –Ј–∞–Љ–µ–і–ї—П—П –Њ–±—Й—Г—О —Б–Ї–Њ—А–Њ—Б—В—М –±–µ–ї–Ї–Њ–≤–Њ–≥–Њ —Б–Є–љ—В–µ–Ј–∞.
–Я–Њ–Љ–Є–Љ–Њ —Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є —В–†–Э–Ъ, Mg2+ —В–∞–Ї–ґ–µ —Б—В–∞–±–Є–ї–Є–Ј–Є—А—Г–µ—В –љ–µ–±–Њ–ї—М—И–Є–µ —П–і–µ—А–љ—Л–µ –†–Э–Ъ (—В.–љ. «snRNA»), –Ї–Њ—В–Њ—А—Л–µ —Г—З–∞—Б—В–≤—Г—О—В –≤ –њ—А–µ–—Б–њ–ї–∞–є—Б–Є–љ–≥–µ –Ї–Њ–і–Є—А—Г—О—Й–µ–є –†–Э–Ъ –љ–∞ —Б–њ–ї–∞–є—Б–µ–Њ—Б–Њ–Љ–µ. –Ф–µ—Д–Є—Ж–Є—В Mg2+ –і–µ—Б—В–∞–±–Є–ї–Є–Ј–Є—А—Г–µ—В snRNA –Є –Љ–Њ–ґ–µ—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї —З–∞—Б—В–Є—З–љ–Њ–є –і–Є—Б—Б–Њ—Ж–Є–∞—Ж–Є–Є (—А–∞—Б–њ–∞–і—Г) —Б–њ–ї–∞–є—Б–µ–Њ—Б–Њ–Љ—Л –љ–∞ –†–Э–Ъ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В—Л –Є –±–µ–ї–Ї–Њ–≤—Л–µ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В—Л. –°—Л–≤–Њ—А–Њ—В–Ї–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б —Б–Є—Б—В–µ–Љ–љ—Л–Љ–Є —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є–Љ–Є –±–Њ–ї–µ–Ј–љ—П–Љ–Є —З–∞—Б—В–Њ —Б–Њ–і–µ—А–ґ–Є—В –∞–љ—В–Є—В–µ–ї–∞ –њ—А–Њ—В–Є–≤ —П–і–µ—А–љ—Л—Е –∞–љ—В–Є–≥–µ–љ–Њ–≤ –Є —Б—А–µ–і–Є –љ–Є—Е –±–µ–ї–Ї–Є, –Њ–±—А–∞–Ј—Г—О—Й–Є–µ –Ї–Њ–Љ–њ–ї–µ–Ї—Б—Л —Б snRNA (U1, U2, U4, U5 –Є U6 [55]). –†–µ–Ј—Г–ї—М—В–∞—В–Њ–Љ —З–∞—Б—В–Є—З–љ–Њ–є –і–Є—Б—Б–Њ—Ж–Є–∞—Ж–Є–Є —Б–њ–ї–∞–є—Б–µ–Њ—Б–Њ–Љ—Л —В–∞–Ї–ґ–µ –±—Г–і–µ—В –Ј–∞–Љ–µ–і–ї–µ–љ–Є–µ —Б–Є–љ—В–µ–Ј–∞ –±–µ–ї–Ї–∞. –°–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ, –і–µ—Д–Є—Ж–Є—В –Љ–∞–≥–љ–Є—П –≤ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –њ—А–Є–≤–µ–і–µ—В –Ї –Ј–∞–Љ–µ–і–ї–µ–љ–Є—О —Б–Є–љ—В–µ–Ј–∞ –≤—Б–µ—Е —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї (–≤–Ї–ї—О—З–∞—П –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ—Л, –≥–ї—О–Ї–Њ–Ј–∞–Љ–Є–љ–Њ–≥–ї–Є–Ї–∞–љ—Л, –Ї–Њ–ї–ї–∞–≥–µ–љ—Л –Є —Н–ї–∞—Б—В–Є–љ). –Я–Њ—Б–Ї–Њ–ї—М–Ї—Г —Б–Є–љ—В–µ–Ј —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї, —Б—В–Њ–ї—М –љ–µ–Њ–±—Е–Њ–і–Є–Љ—Л—Е –і–ї—П –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П («—А–µ–Љ–Њ–љ—В–∞») —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є, –Ј–∞–Љ–µ–і–ї—П–µ—В—Б—П, —В–Њ –њ—А–Њ—Ж–µ—Б—Б—Л –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П —В–∞–Ї–ґ–µ —В–Њ—А–Љ–Њ–Ј—П—В—Б—П, –Є —Н—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї —Г—Е—Г–і—И–µ–љ–Є—О –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Є—Е —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї —В–Ї–∞–љ–Є.
–°–Њ–≤–Љ–µ—Б—В–љ–Њ–µ —А–∞—Б—Б–Љ–Њ—В—А–µ–љ–Є–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ–є –±–Є–Њ–ї–Њ–≥–Є–Є –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж—Л –Є —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ –≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ –Љ–∞–≥–љ–Є—П –Љ–Њ–ґ–µ—В –њ–Њ–Ј–≤–Њ–ї–Є—В—М –љ–∞–Љ —Б—Д–Њ—А–Љ—Г–ї–Є—А–Њ–≤–∞—В—М –±–Њ–ї–µ–µ —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л, —З–µ—А–µ–Ј –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–ґ–µ—В –Њ—Б—Г—Й–µ—Б—В–≤–ї—П—В—М—Б—П –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –Ф–°–Ґ –Є –і–µ—Д–Є—Ж–Є—В–∞ –Љ–∞–≥–љ–Є—П. –Ф–ї—П —В–Њ–≥–Њ —З—В–Њ–±—Л –љ–∞–є—В–Є –њ–Њ–і–Њ–±–љ–Њ–≥–Њ —А–Њ–і–∞ –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є –љ–∞ —Г—А–Њ–≤–љ–µ –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е –≥–µ–љ–Њ–≤, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ —А–∞—Б—Б–Љ–Њ—В—А–µ—В—М –Ї–∞–ґ–і—Л–є –Є–Ј –Њ—Б–љ–Њ–≤–љ—Л—Е —Н–ї–µ–Љ–µ–љ—В–Њ–≤ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж—Л –≤ –Њ—В–і–µ–ї—М–љ–Њ—Б—В–Є.
–Ю—Б–љ–Њ–≤–љ—Л–µ —Н–ї–µ–Љ–µ–љ—В—Л –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж—Л – —Н—В–Њ –≥–µ–ї–µ–Њ–±—А–∞–Ј–љ–∞—П —Б—А–µ–і–∞ —Д–Њ—А–Љ–Є—А—Г–µ–Љ–∞—П –њ—А–Њ—В–µ–Њ–≥–ї–Є–Ї–∞–љ–∞–Љ–Є, —Б—В—А—Г–Ї—В—Г—А–љ—Л–Љ–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–Љ–Є –≤–Њ–ї–Њ–Ї–љ–∞–Љ–Є, —Б–µ—В—З–∞—В—Л–Љ–Є (—А–µ—В–Є–Ї—Г–ї—П—А–љ—Л–Љ–Є) –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–Љ–Є –≤–Њ–ї–Њ–Ї–љ–∞–Љ–Є, –≥–Є–±–Ї–Є–Љ–Є (—Н–ї–∞—Б—В–Є–љ–Њ–≤—Л–Љ–Є) –≤–Њ–ї–Њ–Ї–љ–∞–Љ–Є –Є –Ї–ї–µ—В–Ї–∞–Љ–Є (—Д–Є–±—А–Њ–±–ї–∞—Б—В—Л –Є–ї–Є —Е–Њ–љ–і—А–Њ–±–ї–∞—Б—В—Л).
–Э–Є–Ї–∞–Ї–Є—Е –Њ—В—З–µ—В–ї–Є–≤—Л—Е —Н—Д—Д–µ–Ї—В–Њ–≤ Mg2+ –љ–∞ —Б—В—А—Г–Ї—В—Г—А—Г –≥–µ–ї–µ–Њ–±—А–∞–Ј–љ–Њ–є —Б—Г–±—Б—В–∞–љ—Ж–Є–Є –Т–Ъ–Ь –њ–Њ–Ї–∞ –љ–µ –±—Л–ї–Њ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ. –Т—Б–µ –ґ–µ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ,—З—В–Њ –Є–Њ–љ—Л Mg2+ –Љ–Њ–≥—Г—В –Љ–Њ–і—Г–ї–Є—А–Њ–≤–∞—В—М –∞–Ї—В–Є–≤–љ–Њ—Б—В—М —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–Є—Е –±–Є–Њ—Б–Є–љ—В–µ—В–Є—З–µ—Б–Ї–Є—Е —Д–µ—А–Љ–µ–љ—В–Њ–≤. –Э–∞–њ—А–Є–Љ–µ—А, –≥–Є–∞–ї—Г—А–Њ–љ–∞–љ—Б–Є–љ—В–µ—В–∞–Ј—Л HAS1, HAS2 –Є HAS3 —Б–Њ–і–µ—А–ґ–∞—В –Є–Њ–љ –Љ–∞–≥–љ–Є—П –≤ –∞–Ї—В–Є–≤–љ–Њ–Љ —Ж–µ–љ—В—А–µ (—А–Є—Б. 5). –° –і—А—Г–≥–Њ–є —Б—В–Њ—А–Њ–љ—Л, –Є–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –і–µ–є—Б—В–≤–Є–µ –Є–љ–≥–Є–±–Є—В–Њ—А–Њ–≤ –≥–Є–∞–ї—Г—А–Њ–љ–Є–і–∞–Ј (—Д–µ—А–Љ–µ–љ—В–Њ–≤, –і–µ–≥—А–∞–і–Є—А—Г—О—Й–Є—Е –≥–Є–∞–ї—Г—А–Њ–љ–∞–љ) –Ј–∞–≤–Є—Б–Є—В –Њ—В –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Є–Њ–љ–Њ–≤ –Љ–∞–≥–љ–Є—П [56]. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –і–µ—Д–Є—Ж–Є—В –Љ–∞–≥–љ–Є—П –Љ–Њ–ґ–µ—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї –њ–Њ–љ–Є–ґ–µ–љ–Є—О –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –≥–Є–∞–ї—Г—А–Њ–љ–∞–љ—Б–Є–љ—В–µ—В–∞–Ј –Є –≤ —В–Њ –ґ–µ –≤—А–µ–Љ—П – –Ї –њ–Њ–≤—Л—И–µ–љ–Є—О –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –≥–Є–∞–ї—Г—А–Њ–љ–Є–і–∞–Ј (—В–∞–Ї –Ї–∞–Ї –Є–љ–≥–Є–±–Є—В–Њ—А—Л –њ–µ—А–µ—Б—В–∞—О—В –і–µ–є—Б—В–≤–Њ–≤–∞—В—М –њ—А–Є –љ–µ–і–Њ—Б—В–∞—В–Ї–µ –Љ–∞–≥–љ–Є—П). –Ю–±–∞ —Н—В–Є –њ—А–Њ—Ж–µ—Б—Б–∞ –њ—А–Є–≤–µ–і—Г—В –Ї —Г—Е—Г–і—И–µ–љ–Є—О –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Є—Е —Б–≤–Њ–є—Б—В–≤ –љ–Є—В–µ–є –≥–Є–∞–ї—Г—А–Њ–љ–∞–љ–∞ –Є —З–∞—Б—В–Є—З–љ–Њ–є –і–µ–≥—А–∞–і–∞—Ж–Є–Є –≥–µ–ї—П, –Њ–±—А–∞–Ј—Г—О—Й–µ–≥–Њ –Њ—Б–љ–Њ–≤—Г –Т–Ъ–Ь.
–Ъ–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞ —П–≤–ї—П—О—В—Б—П –Њ—Б–љ–Њ–≤–љ–Њ–є —Б—В—А—Г–Ї—В—Г—А–љ–Њ–є –њ–Њ–і–і–µ—А–ґ–Ї–Њ–є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ш–Ј–±—Л—В–Њ–Ї –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ –Є–ї–Є —Б–ї–Є—И–Ї–Њ–Љ –Љ–∞–ї–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ї–Њ–ї–ї–∞–≥–µ–љ–∞–Ј –њ—А–Є–≤–Њ–і–Є—В –Ї —Г–≤–µ–ї–Є—З–µ–љ–Є—О –њ–ї–Њ—В–љ–Њ—Б—В–Є –≤–Њ–ї–Њ–Ї–Њ–љ –Є –Ї —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—О –Љ–µ–љ–µ–µ –≥–Є–±–Ї–Њ–є —В–Ї–∞–љ–Є. –Э–∞–Њ–±–Њ—А–Њ—В, —З—А–µ–Ј–Љ–µ—А–љ–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ї–Њ–ї–ї–∞–≥–µ–љ–∞–Ј –њ—А–Є–≤–µ–і–µ—В –Ї –љ–µ—Г–њ—А–∞–≤–ї—П–µ–Љ–Њ–є —Д—А–∞–≥–Љ–µ–љ—В–∞—Ж–Є–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞, —З—В–Њ —Б–і–µ–ї–∞–µ—В —В–Ї–∞–љ—М –±–Њ–ї–µ–µ –∞–Љ–Њ—А—Д–љ–Њ–є.
–Э–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ –Є–Њ–љ—Л Mg2+ –љ–µ –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г—О—В –љ–Є —Б –Љ–Њ–ї–µ–Ї—Г–ї–∞–Љ–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞, –љ–Є —Б TIMP –±–µ–ї–Ї–∞–Љ–Є, —В–∞–Ї —З—В–Њ —Н—Д—Д–µ–Ї—В Mg2+ –љ–∞ –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ї–Њ–ї–ї–∞–≥–µ–љ–∞–Ј –Њ—Б–Њ–±–µ–љ–љ–Њ –Є–љ—В–µ—А–µ—Б–µ–љ. –Ш–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –ї–µ—З–µ–љ–Є–µ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Њ—Б—В—А—Л–Љ –Є–љ—Д–∞—А–Ї—В–Њ–Љ –Љ–Є–Њ–Ї–∞—А–і–∞ –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ —Б—Г–ї—М—Д–∞—В–∞ –Љ–∞–≥–љ–Є—П –Є –Њ—А–Њ—В–∞—В–∞ –Љ–∞–≥–љ–Є—П —В–Њ—А–Љ–Њ–Ј–Є—В –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –Љ–Є–Њ–Ї–∞—А–і–∞. –Ю–±—Л—З–љ–Њ –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ—Л–µ —Г—А–Њ–≤–љ–Є IL6 –Є MMP1 –≤ –Ї—А–Њ–≤–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –≤–Њ–Ј—А–∞—Б—В–∞—О—В –њ—А–Є –Њ—Б—В—А–Њ–Љ –Є–љ—Д–∞—А–Ї—В–µ –Љ–Є–Њ–Ї–∞—А–і–∞, –љ–Њ —Н—В–Є —Г—А–Њ–≤–љ–Є –Њ—Б—В–∞—О—В—Б—П –љ–∞ —Б—А–∞–≤–љ–Є—В–µ–ї—М–љ–Њ –љ–Є–Ј–Ї–Њ–Љ —Г—А–Њ–≤–љ–µ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –њ–Њ—Б–ї–µ –Љ–∞–≥–љ–Є–є–—В–µ—А–∞–њ–Є–Є. –Т–Њ–Ј—А–∞—Б—В–∞—О—Й–∞—П –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П IL6 –Љ–Њ–ґ–µ—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї –њ–Њ–≤—Л—И–µ–љ–Є—О –Њ–±—Й–µ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є MMP1, –≤–µ–і—П –Ї –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О —В–Ї–∞–љ–µ–є, –≤ —В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є Mg2+ –≤ —Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є —Г–Љ–µ–љ—М—И–∞–µ—В —Г—А–Њ–≤–љ–Є IL6 –Є MMP1 [57]. –Ф–Њ–њ–Њ–ї–љ–µ–љ–Є–µ –і–Є–µ—В—Л —Д–Њ–ї–Є–µ–≤–Њ–є –Ї–Є—Б–ї–Њ—В–Њ–є –Є —Б–Њ–ї—П–Љ–Є –Љ–∞–≥–љ–Є—П —Г–Љ–µ–љ—М—И–∞–µ—В MMP2 —Б–µ–Ї—А–µ—Ж–Є—О –Є –Њ–Ї–∞–Ј—Л–≤–∞–µ—В –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ–µ –≤–ї–Є—П–љ–Є–µ –љ–∞ –Ш–С–° [58].
–≠–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В—Л –љ–∞ –ґ–Є–≤–Њ—В–љ—Л—Е –њ–Њ–і—В–≤–µ—А–ґ–і–∞—О—В –≤–ї–Є—П–љ–Є–µ –Љ–∞–≥–љ–Є—П –љ–∞ –±–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ь–Ь–Я. –£ –Љ—Л—И–µ–є —Б –Є—Б–Ї—Г—Б—Б—В–≤–µ–љ–љ–Њ –≤—Л–Ј–≤–∞–љ–љ—Л–Љ –і–µ—Д–Є—Ж–Є—В–Њ–Љ –Љ–∞–≥–љ–Є—П —Б—В–µ–љ–Ї–∞ –∞–Њ—А—В—Л –±—Л–ї–∞ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —В–Њ–љ—М—И–µ, —З–µ–Љ —Г –Ї–Њ–љ—В—А–Њ–ї—М–љ—Л—Е –ґ–Є–≤–Њ—В–љ—Л—Е. –°–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–∞—П –Њ–Ї—А–∞—Б–Ї–∞ –і–≤—Г—Е –Њ—Б–љ–Њ–≤–љ—Л—Е —В–Є–њ–Њ–≤ –≤–Њ–ї–Њ–Ї–Њ–љ (–Ї–Њ–ї–ї–∞–≥–µ–љ–∞ –Є —Н–ї–∞—Б—В–Є–љ–∞) –њ–Њ–Ї–∞–Ј–∞–ї–∞ —Б—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–µ —Б—В—А—Г–Ї—В—Г—А–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Њ–±–Њ–Є—Е –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤. –≠—В–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Ї–Њ—А–µ–ї–ї–Є—А–Њ–≤–∞–ї–Є —Б –њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ –Њ–±—Й–µ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Љ–∞—В—А–Є—З–љ—Л—Е –Љ–µ—В–∞–ї–ї–Њ–њ—А–Њ—В–µ–Є–љ–∞–Ј MMP2 –Є MMP9 [59]. –Т –Ї–ї–µ—В–Ї–∞—Е –≥–ї–∞–і–Ї–Њ–є –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А—Л —Б–Њ—Б—Г–і–Њ–≤ —Г –Ї—А—Л—Б –і–Њ–±–∞–≤–ї–µ–љ–Є–µ –Љ–∞–≥–љ–Є—П —Г–Љ–µ–љ—М—И–∞–ї–Њ –Њ–±—Й—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М MMP2 –њ—А—П–Љ–Њ –њ—А–Њ–њ–Њ—А—Ж–Є–Њ–љ–∞–ї—М–љ–Њ –і–Њ–Ј–µ –Љ–∞–≥–љ–Є—П [60]. –Т–Њ–Ј–Љ–Њ–ґ–љ–Њ, —З—В–Њ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–Є–є —Б–Є–≥–љ–∞–ї—М–љ—Л–є –Ї–∞—Б–Ї–∞–і –Ј–∞–њ—Г—Б–Ї–∞–µ—В—Б—П –Њ–і–љ–Є–Љ –Є–Ј –±–µ–ї–Ї–Њ–≤–—Б–µ–љ—Б–Њ—А–Њ–≤ –Љ–∞–≥–љ–Є—П (–љ–∞–њ—А–Є–Љ–µ—А, CASR, TRPM7, —Б–Љ. –≤—Л—И–µ).
–Ф–∞–љ–љ—Л–µ, –њ—А–Є–≤–µ–і–µ–љ–љ—Л–µ –≤—Л—И–µ, –њ–Њ–Ј–≤–Њ–ї—П—О—В –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В—М, —З—В–Њ –і–µ—Д–Є—Ж–Є—В Mg2+ –і–Њ–ї–ґ–µ–љ, –≤–µ—А–Њ—П—В–љ–Њ, –њ—А–Є–≤–Њ–і–Є—В—М –Ї –њ–Њ–≤—Л—И–µ–љ–Є—О –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ь–Ь–Я (–≤ —З–∞—Б—В–љ–Њ—Б—В–Є, –Ї–Њ–ї–ї–∞–≥–µ–љ–∞–Ј), –Ї–Њ—В–Њ—А—Л–µ –љ–∞—З–Є–љ–∞—О—В –і–µ–≥—А–∞–і–Є—А–Њ–≤–∞—В—М —Б—В—А—Г–Ї—В—Г—А–љ—Л–µ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В—Л –Т–Ъ–Ь (–њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –Ї–Њ–ї–ї–∞–≥–µ–љ) —Б –±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Њ–є —Б–Ї–Њ—А–Њ—Б—В—М—О. –≠—В–Є —Н—Д—Д–µ–Ї—В—Л –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –Љ–∞–≥–љ–Є—П, –љ–∞–Є–±–Њ–ї–µ–µ –≤–µ—А–Њ—П—В–љ–Њ, –≤—Л–Ј—Л–≤–∞—О—В—Б—П —З–µ—А–µ–Ј –љ–µ–Ї–Є–є, –њ–Њ–Ї–∞ –љ–µ –Є–Ј–≤–µ—Б—В–љ—Л–є –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ—Л–є —Б–Є–≥–љ–∞–ї—М–љ—Л–є –Ї–∞—Б–Ї–∞–і. –Э–µ–ї—М–Ј—П –Є—Б–Ї–ї—О—З–∞—В—М —В–∞–Ї–ґ–µ –Є –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—М –њ—А—П–Љ–Њ–≥–Њ –Є–љ–≥–Є–±–Є—А–Њ–≤–∞–љ–Є—П —А–∞–Ј–ї–Є—З–љ—Л—Е –Ь–Ь–Я –Є–Њ–љ–∞–Љ–Є –Љ–∞–≥–љ–Є—П —З–µ—А–µ–Ј –Ї–Њ–љ–Ї—Г—А–µ–љ—В–љ–Њ–µ —Б–≤—П–Ј—Л–≤–∞–љ–Є–µ –і–≤—Г—Е–≤–∞–ї–µ—В–љ—Л—Е –Ї–∞—В–Є–Њ–љ–Њ–≤ –≤ –∞–Ї—В–Є–≤–љ–Њ–Љ —Ж–µ–љ—В—А–µ (—А–Є—Б. 5) –Є–ї–Є —З–µ—А–µ–Ј –∞–ї–ї–Њ—Б—В–µ—А–Є—З–µ—Б–Ї–Є–µ –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є—П —Б –Ь–Ь–Я.
–≠—Д—Д–µ–Ї—В—Л Mg2+ –љ–∞ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ—Г—О —В–Ї–∞–љ—М –љ–µ –Њ–≥—А–∞–љ–Є—З–Є–≤–∞—О—В—Б—П –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–Љ –Є –Ї–Њ–ї–ї–∞–≥–µ–љa–Ј–∞–Љ–Є. –Ь–Є–Ї—А–Њ—Д–Є–±—А–Є–ї—Л –Є —Н–ї–∞—Б—В–Є–љ – –Њ—Б–љ–Њ–≤–љ—Л–µ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В—Л –≥–Є–±–Ї–Є—Е –≤–Њ–ї–Њ–Ї–Њ–љ. –Ф–µ–≥—А–∞–і–∞—Ж–Є—П –≤–Њ–ї–Њ–Ї–Њ–љ —Н–ї–∞—Б—В–Є–љ–∞ –Љ–Њ–ґ–µ—В –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –≤–Њ–Ј—А–∞—Б—В–∞—В—М (–≤ 2–3 —А–∞–Ј–∞) –≤ –њ—А–Є—Б—Г—В—Б—В–≤–Є–Є Mg2+ [61]. –Ф–µ—Д–Є—Ж–Є—В Mg2+ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В –±–Њ–ї–µ–µ –љ–Є–Ј–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —Н–ї–∞—Б—В–∞–Ј –Є –±–Њ–ї—М—И–µ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –≥–Є–±–Ї–Є—Е –≤–Њ–ї–Њ–Ї–Њ–љ. –Т –Њ—Б–љ–Њ–≤–љ–Њ–Љ, –Њ–і–љ–∞–Ї–Њ, —Б–Ї–Њ—А–µ–µ, –Ї–∞–ї—М—Ж–Є–µ–≤—Л–µ, –∞ –љ–µ –Љ–∞–≥–љ–Є–µ–≤—Л–µ –Є–Њ–љ—Л –≤–ї–Є—П—О—В –љ–∞ –≥–Є–±–Ї–Є–µ –≤–Њ–ї–Њ–Ї–љ–∞: Ca2+ –љ–µ–Њ–±—Е–Њ–і–Є–Љ –і–ї—П –∞–Ї—В–Є–≤–љ—Л—Е —Ж–µ–љ—В—А–Њ–≤ —Н–ї–∞—Б—В–∞–Ј, Ca2+ —В–∞–Ї–ґ–µ —Б—В–∞–±–Є–ї–Є–Ј–Є—А—Г–µ—В —Б—В—А—Г–Ї—В—Г—А—Г –Љ–Є–Ї—А–Њ—Д–Є–±—А–Є–ї (–≤ —З–∞—Б—В–љ–Њ—Б—В–Є, —З–µ—А–µ–Ј –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є—П —Б —Д–Є–±—А–Є–ї–ї–Є–љ–Њ–Љ–1 [62] –Є –Љ–Є–Ї—А–Њ—Д–Є–±—А–Є–ї–—Б–≤—П–Ј–∞–љ–љ—Л–Љ –≥–ї–Є–Ї–Њ–±–µ–ї–Ї–Њ–Љ–1 (MAGP–1) [63]).
–Ґ—А–∞–љ—Б–≥–ї—Г—В–∞–Љ–Є–љ–∞–Ј–∞ – —Д–µ—А–Љ–µ–љ—В, —Д–Њ—А–Љ–Є—А—Г—О—Й–Є–є –њ–Њ–њ–µ—А–µ—З–љ—Л–µ –≥–ї—Г—В–∞–Љ–Є–љ––ї–Є–Ј–Є–љ–Њ–≤—Л–µ —Б—И–Є–≤–Ї–Є, —Б–Њ–µ–і–Є–љ—П—О—Й–Є–µ –≤–Љ–µ—Б—В–µ —Ж–µ–њ–Є —Н–ї–∞—Б—В–Є–љ–∞, –∞–Ї—В–Є–≤–Є–Ј–Є—А—Г–µ—В—Б—П Ca2+ –Є –Є–љ–≥–Є–±–Є—А—Г–µ—В—Б—П Mg2+ [64,65]. Mg2+ –Љ–Њ–ґ–µ—В –Є–љ–≥–Є–±–Є—А–Њ–≤–∞—В—М –Љ–µ–і—М––Ј–∞–≤–Є—Б–Є–Љ—Г—О –ї–Є–Ј–Є–ї–Њ–Ї—Б–Є–і–∞–Ј—Г (LOX) [66], —В–∞–Ї–ґ–µ –≤–Њ–≤–ї–µ—З–µ–љ–љ—Г—О –≤ –њ–Њ–њ–µ—А–µ—З–љ—Г—О —Б—И–Є–≤–Ї—Г —Ж–µ–њ–µ–є —Н–ї–∞—Б—В–Є–љ–Њ–≤ –Є/–Є–ї–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤. –°–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ, –і–µ—Д–Є—Ж–Є—В Mg2+ –Љ–Њ–ґ–µ—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї –∞–Ї—В–Є–≤–Є–Ј–∞—Ж–Є–Є –њ–Њ–њ–µ—А–µ—З–љ–Њ–є —Б—И–Є–≤–Ї–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤ –Є —Н–ї–∞—Б—В–Є–љ–Њ–≤, –Є —Н—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б, –љ–∞—А—П–і—Г —Б —Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ь–Ь–Я, –њ—А–Є–≤–µ–і–µ—В –Ї —Б–≤–Њ–µ–≥–Њ —А–Њ–і–∞ –≥—А–∞–љ—Г–ї—П—А–Є–Ј–∞—Ж–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є.
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Є–Љ–µ—О—Й–Є–µ—Б—П –і–∞–љ–љ—Л–µ –њ–Њ–Ј–≤–Њ–ї—П—О—В —Б–і–µ–ї–∞—В—М –≤—Л–≤–Њ–і, —З—В–Њ –љ–∞–Є–±–Њ–ї–µ–µ –≤–µ—А–Њ—П—В–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –і–µ—Д–Є—Ж–Є—В–∞ Mg2+ –љ–∞ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ—Г—О —В–Ї–∞–љ—М – —Н—В–Њ —Г—Б–Є–ї–µ–љ–Є–µ –і–µ–≥—А–∞–і–∞—Ж–Є–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л—Е –Є, –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, —Н–ї–∞—Б—В–Є–љ–Њ–≤—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ, –∞ —В–∞–Ї–ґ–µ –њ–Њ–ї–Є—Б–∞—Е–∞—А–Є–і–љ—Л—Е –љ–Є—В–µ–є –≥–Є–∞–ї—Г—А–Њ–љ–∞–љ–∞ (—А–Є—Б. 6). –Э–µ–ї—М–Ј—П –Є—Б–Ї–ї—О—З–Є—В—М, —З—В–Њ —Г—Б–Є–ї–µ–љ–Є–µ –њ–Њ–њ–µ—А–µ—З–љ—Л—Е —Б—И–Є–≤–Њ–Ї –њ—А–Є–≤–µ–і–µ—В –Ї –≥—А–∞–љ—Г–ї—П—А–Є–Ј–∞—Ж–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є, —А–∞—Б—Б–ї–Њ–µ–љ–Є—О –љ–∞ —Н—В–∞–Ї–Є–µ «–њ–ї–∞—Б—В–Є–љ–Ї–Є», —Б–Њ—Б—В–Њ—П—Й–Є–µ –Є–Ј –љ–∞–њ–Њ–ї–Њ–≤–Є–љ—Г –і–µ–≥—А–∞–і–Є—А–Њ–≤–∞–љ–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤, –Є –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ –њ—А–Є–≤–Њ–і–Є—В –Ї —Г–Љ–µ–љ—М—И–µ–љ–Є—О –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Њ–є –њ—А–Њ—З–љ–Њ—Б—В–Є. –Я—А–Є –і–Њ—Б—В–∞—В–Њ—З–љ–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є Mg2+ —Б–µ–Ї—А–µ—Ж–Є—П/–∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ь–Ь–Я —Б–љ–Є–ґ–∞—О—В—Б—П, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї —Г–Љ–µ–љ—М—И–µ–љ–Є—О –і–µ–≥—А–∞–і–∞—Ж–Є–Є –Є –Ї —Г—Б–Ї–Њ—А–µ–љ–Є—О –±–µ–ї–Ї–Њ–≤–Њ–≥–Њ —Б–Є–љ—В–µ–Ј–∞ –љ–Њ–≤—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤.
–І–∞—Б—В–љ—Л–є —Б–ї—Г—З–∞–є:
Bw35 –∞–љ—В–Є–≥–µ–љ (–≥–µ–љ HLA–B)
–Я—А–Є–≤–µ–і–µ–љ–љ—Л–є –≤—Л—И–µ –∞–љ–∞–ї–Є–Ј –Њ—В–і–µ–ї—М–љ—Л—Е –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–є –Љ–∞—В—А–Є—Ж—Л –њ—А–µ–і–Њ—Б—В–∞–≤–Є–ї –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—М —Б–Є—Б—В–µ–Љ–∞—В–Є–Ј–Є—А–Њ–≤–∞—В—М –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –і–µ—Д–Є—Ж–Є—В–∞ –Љ–∞–≥–љ–Є—П –Є –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –°—Г—Й–µ—Б—В–≤—Г–µ—В, —В–µ–Љ –љ–µ –Љ–µ–љ–µ–µ, –µ—Й–µ –Њ–і–Є–љ –Ї–Њ–љ–Ї—А–µ—В–љ—Л–є —Б–ї—Г—З–∞–є, –Ї–Њ—В–Њ—А—Л–є –Љ–Њ–ґ–µ—В –Њ–±—Г—Б–ї–Њ–≤–ї–Є–≤–∞—В—М –Ф–°–Ґ, –љ–µ –Є–Љ–µ—О—Й–Є–є, –Њ–і–љ–∞–Ї–Њ, –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ–≥–Њ –Њ—В–љ–Њ—И–µ–љ–Є—П –Ї —Б—В—А—Г–Ї—В—Г—А–µ –Т–Ъ–Ь. –†–µ—З—М –Є–і–µ—В –Њ –Ї—А–∞–є–љ–µ –Є–љ—В–µ—А–µ—Б–љ—Л—Е –і–∞–љ–љ—Л—Е –њ–Њ –∞–љ—В–Є–≥–µ–љ—Г Bw35.
–Ш–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –њ–∞—Ж–Є–µ–љ—В—Л —Б –њ—А–Њ–ї–∞–њ—Б–Њ–Љ –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ —З–∞—Б—В–Њ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П –њ—А–Є—Б—Г—В—Б—В–≤–Є–µ–Љ –∞–љ—В–Є–≥–µ–љ–∞ Bw35 —Б–Є—Б—В–µ–Љ—Л HLA (–ї–Є–Љ—Д–Њ—Ж–Є—В–∞—А–љ—Л–є –∞–љ—В–Є–≥–µ–љ –≥–Є—Б—В–Њ—Б–Њ–≤–Љ–µ—Б—В–Є–Љ–Њ—Б—В–Є, —З–∞—Б—В—М MHC —Б–Є—Б—В–µ–Љ—Л –Є–Љ–Љ—Г–љ–љ–Њ–є –Ј–∞—Й–Є—В—Л) –Ї–Њ—В–Њ—А—Л–є —В–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ (—З–µ—А–µ–Ј —Д–∞–Ї—В –њ—А–Є—Б—Г—В—Б—В–≤–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П) –∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П —Б –∞–±–љ–Њ—А–Љ–∞–ї—М–љ—Л–Љ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–Њ–Љ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є [1,67] –Є —Б –Љ–∞–≥–љ–Є–є––і–µ—Д–Є—Ж–Є—В–Њ–Љ [1,68,69]. –Р–ї–ї–µ–ї—М Bw35 —Б–Є—Б—В–µ–Љ—Л HLA, –љ–∞—А—П–і—Г —Б –њ–Њ–≤–µ–і–µ–љ—З–µ—Б–Ї–Є–Љ —В–Є–њ–Њ–Љ «–Р» (–∞–≥—А–µ—Б—Б–Є–≤–љ—Л–є, –љ–µ—Г—А–∞–≤–љ–Њ–≤–µ—И–µ–љ–љ—Л–є —В–Є–њ –њ–Њ–≤–µ–і–µ–љ–Є—П), –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є –Ї–Њ–љ—Б—В–Є—В—Г—Ж–Є–Њ–љ–љ—Л–є —Д–∞–Ї—В–Њ—А, —Г–Ї–∞–Ј—Л–≤–∞—О—Й–Є–є –љ–∞ –њ–Њ–≤—Л—И–µ–љ–љ—Г—О –њ–Њ—В—А–µ–±–љ–Њ—Б—В—М –≤ –Љ–∞–≥–љ–Є–Є [70].
–†–µ—Ж–µ–њ—В–Њ—А MHC –Њ—Б–љ–Њ–≤–љ–Њ–≥–Њ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –≥–Є—Б—В–Њ—Б–Њ–≤–Љ–µ—Б—В–Є–Љ–Њ—Б—В–Є —В–Є–њ–∞ 1 –Є–≥—А–∞–µ—В —Ж–µ–љ—В—А–∞–ї—М–љ—Г—О —А–Њ–ї—М –≤ –Є–Љ–Љ—Г–љ–љ–Њ–є —Б–Є—Б—В–µ–Љ–µ, –њ—А–µ–Ј–µ–љ—В–Є—А—Г—П –њ–µ–њ—В–Є–і––њ—А–Њ–Є–Ј–≤–Њ–і–љ—Л–µ –±–µ–ї–Ї–Њ–≤ —Ж–Є—В–Њ–Ј–Њ–ї—П –Є —Н–љ–і–Њ–њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Њ–≥–Њ —А–µ—В–Є–Ї—Г–ї—Г–Љ–∞ –љ–∞ –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–Є –Ї–ї–µ—В–Њ–Ї. –Ґ–∞–Ї–Є–µ —А–µ—Ж–µ–њ—В–Њ—А—Л —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г—О—В—Б—П –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –≤ –Ї–∞–ґ–і–Њ–є –Ї–ї–µ—В–Ї–µ, –Є T––ї–Є–Љ—Д–Њ—Ж–Є—В—Л –∞—В–∞–Ї—Г—О—В —В–µ –Ї–ї–µ—В–Ї–Є, MHC —А–µ—Ж–µ–њ—В–Њ—А—Л –Ї–Њ—В–Њ—А—Л—Е –њ—А–µ–Ј–µ–љ—В–Є—А—Г—О—В —З—Г–ґ–µ—А–Њ–і–љ—Л–µ –њ–µ–њ—В–Є–і—Л. –Т–Њ–Ј–Љ–Њ–ґ–љ–Њ, —З—В–Њ –Ґ––ї–Є–Љ—Д–Њ—Ж–Є—В—Л –±—Г–і—Г—В –∞—В–∞–Ї–Њ–≤–∞—В—М –Ї–ї–µ—В–Ї–Є, —Б–Њ–і–µ—А–ґ–∞—Й–Є–µ –Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л–µ –Љ—Г—В–Є—А–Њ–≤–∞–љ–љ—Л–µ —Д–Њ—А–Љ—Л MHC —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ –њ—А–Є–Ј–љ–∞—О—В—Б—П –Ґ––ї–Є–Љ—Д–Њ—Ж–Є—В–∞–Љ–Є –Ї–∞–Ї «—З—Г–ґ–µ—А–Њ–і–љ—Л–µ». –≠—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б, –Њ—З–µ–≤–Є–і–љ–Њ, –≤—Л–Ј–Њ–≤–µ—В –њ–Њ–≤—Л—И–µ–љ–Є–µ –∞—Г—В–Њ–Є–Љ—Г–љ–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є. –У–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –і–µ—Д–µ–Ї—В—Л –≥–µ–љ–∞ HLA–B –Љ–Њ–≥—Г—В –њ—А–Є–≤–µ—Б—В–Є –Ї —Б–њ–Њ–љ–і–Є–ї–Њ–∞—А—В—А–Њ–њ–∞—В–Є–Є, –Њ–і–љ–Њ–Љ—Г –Є–Ј –љ–∞–Є–±–Њ–ї–µ–µ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ—Л—Е —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є—Е —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –∞ —В–∞–Ї–ґ–µ –Ї –і—А—Г–≥–Є–Љ —Б—Е–Њ–і–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ (–љ–∞–њ—А–Є–Љ–µ—А, –∞–љ–Ї–Є–ї–Њ–Ј–љ–Њ–Љ—Г —Б–њ–Њ–љ–і–Є–ї–Є—В—Г [71,72]).
–Т —З–∞—Б—В–љ–Њ—Б—В–Є, –∞–ї–ї–µ–ї—М bw35 –≥–µ–љ–∞ HLA–B –±—Л–ї –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ —Б —В—П–ґ–µ–ї—Л–Љ –њ—А–Њ—В–µ–Ї–∞–љ–Є–µ–Љ –≥–Є–њ–µ—А—В–Њ–љ–Є–Є, —В—П–ґ–µ–ї—Л–Љ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ–Љ —Б–Њ—Б—Г–і–Њ–≤ [73], –њ—Б–Њ—А–Є–∞–Ј–Њ–Љ [74], –њ–Њ–≤—Л—И–µ–љ–љ–Њ–є –≤–Њ—Б–њ—А–Є–Є–Љ—З–Є–≤–Њ—Б—В—М—О –Ї –≥–µ–њ–∞—В–Є—В—Г B [75], –Љ–Є–µ–ї–Њ–Љ–Њ–є [76], —В–Є—А–Њ–Є–і–Є—В–Њ–Љ [77], –∞ —В–∞–Ї–ґ–µ —Б –і—А—Г–≥–Є–Љ–Є –∞—Г—В–Њ–Є–Љ—Г–љ–љ—Л–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ–Є [78], —В–∞–Ї–Є–Љ–Є –Ї–∞–Ї –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–∞—П –±–Њ–ї–µ–Ј–љ—М –Ї–Є—И–µ—З–љ–Є–Ї–∞, —А–µ–≤–Љ–∞—В–Њ–Є–і–љ—Л–є –∞—А—В—А–Є—В –Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–∞—П —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є—П. –Т—Б–µ —Н—В–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Є–Љ–µ—О—В –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–є –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В, —Б–≤—П–Ј–∞–љ–љ—Л–є —Б —Б–Њ—Б—В–Њ—П–љ–Є–µ–Љ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є —Е—А—П—Й–µ–є/—Б–≤—П–Ј–Њ–Ї.
–Т–µ—А–Њ—П—В–љ–Њ–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ–µ –Њ–±—К—П—Б–љ–µ–љ–Є–µ —Н—В–Њ–Љ—Г –Ј–∞–Ї–ї—О—З–∞–µ—В—Б—П –≤ —Б–ї–µ–і—Г—О—Й–µ–Љ: MHC —А–µ—Ж–µ–њ—В–Њ—А—Л, —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–Є–µ –∞–љ—В–Є–≥–µ–љ—Г bw35, –њ—А–Њ–≤–Њ—Ж–Є—А—Г—О—В T––Ї–ї–µ—В–Ї–Є –∞—В–∞–Ї–Њ–≤–∞—В—М —В–Ї–∞–љ–Є, —Б–Њ–і–µ—А–ґ–∞—Й–Є–µ —Н—В—Г —Д–Њ—А–Љ—Г —А–µ—Ж–µ–њ—В–Њ—А–∞. –≠—В–Њ, –≤ —Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –њ—А–Є–≤–Њ–і–Є—В –Ї —Г—Б–Є–ї–µ–љ–љ–Њ–є –∞—Г—В–Њ–Є–Љ—Г–љ–љ–Њ–є —А–µ–∞–Ї—Ж–Є–Є –Є, –Ї–∞–Ї —Б–ї–µ–і—Б—В–≤–Є–µ, –Ї —Г—Б–Є–ї–µ–љ–љ–Њ–є –і–µ–≥—А–∞–і–∞—Ж–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Я–Њ–≤—Л—И–µ–љ–љ–∞—П –і–µ–≥—А–∞–і–∞—Ж–Є—П —В–Ї–∞–љ–Є —З–µ—А–µ–Ј –∞—Г—В–Њ–Є–Љ—Г–љ–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –љ–µ–Є–Ј–±–µ–ґ–љ–Њ –њ—А–Є–≤–µ–і–µ—В –Ї –љ–µ–Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ–Њ–є –њ–Њ—В–µ—А–Є –Љ–∞–≥–љ–Є—П –Є–Ј —В–Ї–∞–љ–Є –Є –Ї —Г–≤–µ–ї–Є—З–µ–љ–Є—О –≤—Л–і–µ–ї–µ–љ–Є—П –Є–Њ–љ–Њ–≤ –Љ–∞–≥–љ–Є—П —З–µ—А–µ–Ј –њ–Њ—З–Ї–Є.
–Ф—А—Г–≥–Њ–µ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ–µ –Њ–±—К—П—Б–љ–µ–љ–Є–µ —Н—В–Њ–≥–Њ —Д–µ–љ–Њ–Љ–µ–љ–∞ –≤–Ї–ї—О—З–∞–µ—В —Е–Њ—А–Њ—И–Њ –Є–Ј–≤–µ—Б—В–љ—Л–є –≤ –≥–µ–љ–µ—В–Є–Ї–µ —Н—Д—Д–µ–Ї—В «–љ–µ—А–∞–≤–љ–Њ–≤–µ—Б–љ–Њ–≥–Њ —Б—Ж–µ–њ–ї–µ–љ–Є—П» (–∞–љ–≥–ї. «linkage disequilibrium»). –Э–µ—А–∞–≤–љ–Њ–≤–µ—Б–љ–Њ–µ —Б—Ж–µ–њ–ї–µ–љ–Є–µ —Г—З–∞—Б—В–Ї–Њ–≤ —Е—А–Њ–Љ–Њ—Б–Њ–Љ—Л –Њ–Ј–љ–∞—З–∞–µ—В, —З—В–Њ –≤–Њ –≤—А–µ–Љ—П –Љ–µ–є–Њ–Ј–∞ –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М —А–µ–Ї–Њ–Љ–±–Є–љ–∞—Ж–Є–Є –і–∞–љ–љ—Л—Е —Г—З–∞—Б—В–Ї–Њ–≤ —З—А–µ–Ј–≤—Л—З–∞–є–љ–Њ –Љ–∞–ї–∞ –Є –і–∞–љ–љ—Л–µ —Г—З–∞—Б—В–Ї–Є —Е—А–Њ–Љ–Њ—Б–Њ–Љ—Л –љ–∞—Б–ї–µ–і—Г—О—В—Б—П –µ–і–Є–љ—Л–Љ –±–ї–Њ–Ї–Њ–Љ. –Т —Н—В–Њ–Љ —Б–ї—Г—З–∞–µ bw35 –∞–ї–ї–µ–ї—М –љ–µ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В –Ї–∞–Ї–Є–Љ –±—Л —В–Њ –љ–Є –±—Л–ї–Њ –∞–љ–Њ–Љ–∞–ї—М–љ—Л–Љ —Д–Њ—А–Љ–∞–Љ MHC —А–µ—Ж–µ–њ—В–Њ—А–∞, –љ–Њ –≤—Л—Б—В—Г–њ–∞–µ—В –≤ –Ї–∞—З–µ—Б—В–≤–µ –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –Љ–∞—А–Ї–µ—А–∞ –і–ї—П –≥–µ–љ–Њ–≤, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –≤ –љ–µ—А–∞–≤–љ–Њ–≤–µ—Б–љ–Њ–Љ —Б—Ж–µ–њ–ї–µ–љ–Є–Є —Б –≥–µ–љ–Њ–Љ HLA–B. –Ъ–∞–Ї –њ–Њ–Ї–∞–Ј—Л–≤–∞–µ—В —Б–Њ–≤–Љ–µ—Б—В–љ—Л–є –∞–љ–∞–ї–Є–Ј –њ–Њ—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—М–љ–Њ—Б—В–Є –Є –≤–∞—А–Є–∞—Ж–Є–є —З–µ–ї–Њ–≤–µ—З–µ—Б–Ї–Њ–≥–Њ –≥–µ–љ–Њ–Љ–∞, —Г –µ–≤—А–Њ–њ–µ–Њ–Є–і–Њ–≤ –Ї–ї–∞—Б—В–µ—А HLA –≥–µ–љ–Њ–≤ (—Ж–Є—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–∞—П –њ–Њ–ї–Њ—Б–∞ 6p21) –≤–Њ–≤–ї–µ—З–µ–љ –≤ –Њ—З–µ–љ—М –±–Њ–ї—М—И—Г—О –≥—А—Г–њ–њ—Г –љ–µ—А–∞–≤–љ–Њ–≤–µ—Б–љ–Њ–≥–Њ —Б—Ж–µ–њ–ї–µ–љ–Є—П (–±–Њ–ї–µ–µ 2 Mb –љ—Г–Ї–ї–µ–Њ—В–Є–і–Њ–≤). –≠—В–∞ –≥—А—Г–њ–њ–∞ —Б—Ж–µ–њ–ї–µ–љ–љ—Л—Е –≥–µ–љ–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ –љ–∞—Б–ї–µ–і—Г—О—В—Б—П, –Ї–∞–Ї –µ–і–Є–љ—Л–є –±–ї–Њ–Ї —Г –µ–≤—А–Њ–њ–µ–Њ–Є–і–Њ–≤, –≤–Ї–ї—О—З–∞–µ—В 14 –≥–µ–љ–Њ–≤. –°—А–µ–і–Є –љ–Є—Е – —В—А–Є –≥–µ–љ–∞ HLA–—Б–Є—Б—В–µ–Љ—Л, –≥–Є–њ–Њ—В–µ—В–Є—З–µ—Б–Ї–Є–є –≥–µ–љ –њ–Њ–і–Њ–±–љ—Л–є –і–Є–≥–Є–і—А–Њ—Д–Њ–ї–∞—В—А–µ–і—Г–Ї—В–∞–Ј—Л (LOC729816), –і–≤–∞ –≥–µ–љ–∞, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Л—Е —Б –≤–Њ—Б–њ—А–Є–Є–Љ—З–Є–≤–Њ—Б—В—М—О –Ї –њ—Б–Њ—А–Є–∞–Ј—Г (PSORS1C1, PSORS1C2), –Ї–Њ—А–љ–µ–Њ–і–µ—Б–Љ–Њ–Ј–Є–љ (CDSN), –≤–Њ–≤–ї–µ—З–µ–љ–љ—Л–є –≤ –і–µ—Б–Ї–≤–∞–Љ–∞—Ж–Є—О —Н–њ–Є—В–µ–ї–Є—П, –Є —В—А–Є –≥–µ–љ–∞, —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б –њ—А–Њ—Ж–µ—Б—Б–∞–Љ–Є –і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–∞–љ–Є—П –Є –Љ–Њ—А—Д–Њ–≥–µ–љ–µ–Ј–∞ –Ї–ї–µ—В–Њ–Ї (CCHCR1, TCF19, POU5F1). –§—Г–љ–Ї—Ж–Є–Є –Њ—Б—В–∞–ї—М–љ—Л—Е –≥–µ–љ–Њ–≤ –њ–Њ–Ї–∞ –љ–µ –Є–Ј–≤–µ—Б—В–љ—Л. –Т –і–∞–љ–љ–Њ–Љ —Б–ї—Г—З–∞–µ bw35 –∞–љ—В–Є–≥–µ–љ HLA–B –Љ–Њ–ґ–µ—В –љ–µ –±—Л—В—М —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–Љ –∞–ї–ї–µ–ї–µ–Љ, –∞ –њ—А–Њ—Б—В–Њ —П–≤–ї—П—В—М—Б—П –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–Љ –Љ–∞—А–Ї–µ—А–Њ–Љ, —Г–Ї–∞–Ј—Л–≤–∞—О—Й–Є–Љ –љ–∞ –њ—А–Є—Б—Г—В—Б—В–≤–Є–µ –∞–±–љ–Њ—А–Љ–∞–ї—М–љ—Л—Е —Д–Њ—А–Љ –ї—О–±–Њ–≥–Њ –Є–Ј —Н—В–Є—Е 14 –≥–µ–љ–Њ–≤.
–Т—Л–≤–Њ–і—Л
–Т–љ–µ –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В —В–Њ–≥–Њ, –≤–Њ–Ј–љ–Є–Ї–∞–µ—В –ї–Є –∞–љ–Њ–Љ–∞–ї–Є—П –≤ —Б—В—А—Г–Ї—В—Г—А–µ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –Є–Ј––Ј–∞ –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ–Њ–≥–Њ –Њ—В–≤–µ—В–∞, –Є–Ј––Ј–∞ –∞–±–љ–Њ—А–Љ–∞–ї—М–љ–Њ–≥–Њ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ —В–Ї–∞–љ–Є –Є —З—А–µ–Ј–Љ–µ—А–љ–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞–Ј –Є–ї–Є –ґ–µ –≤—Б–ї–µ–і—Б—В–≤–Є–µ –і—А—Г–≥–Є—Е –њ—А–Є—З–Є–љ, —Б–Њ—Б—В–Њ—П–љ–Є–µ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є —В–Њ–ї—М–Ї–Њ —Г–ї—Г—З—И–Є—В—Б—П, –µ—Б–ї–Є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞–Ј –Є —Н–ї–∞—Б—В–∞–Ј, –∞ —В–∞–Ї–ґ–µ –±–Є–Њ—Б–Є–љ—В–µ—В–Є—З–µ—Б–Ї–Є—Е —Д–µ—А–Љ–µ–љ—В–Њ–≤ –≥–ї—О–Ї–Њ–Ј–∞–Љ–Є–љ–Њ–≥–ї–Є–Ї–∞–љ–Њ–≤ (–≥–Є–∞–ї—Г—А–Њ–љ–∞–љ—Б–Є–љ—В–µ—В–∞–Ј, –≥–Є–∞–ї—Г—А–Њ–љ–Є–і–∞–Ј, b––≥–∞–ї–∞–Ї—В–Њ–Ј–Є–і–∞–Ј) –±—Г–і—Г—В —Б–±–∞–ї–∞–љ—Б–Є—А–Њ–≤–∞–љ—Л.
–Ф–Њ—Б—В–Є–ґ–µ–љ–Є–µ —В–∞–Ї–Њ–≥–Њ –±–∞–ї–∞–љ—Б–∞, –њ–Њ –≤—Б–µ–є –≤–Є–і–Є–Љ–Њ—Б—В–Є, –і–Њ—Б—В–Є–≥–∞–µ—В—Б—П –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ—Л–Љ —Н—Д—Д–µ–Ї—В–Њ–Љ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –∞–і–µ–Ї–≤–∞—В–љ—Л—Е –і–Њ–Ј –Є–Њ–љ–Њ–≤ –Љ–∞–≥–љ–Є—П. –Э–∞–њ—А–Њ—В–Є–≤, –њ—А–Є –і–µ—Д–Є—Ж–Є—В–µ Mg2+ —Б–Є–љ—В–µ–Ј –±–µ–ї–Ї–Њ–≤ –≤ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –Ј–∞–Љ–µ–і–ї—П–µ—В—Б—П, –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Ь–Ь–Я —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –Є –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–∞—П –Љ–∞—В—А–Є—Ж–∞ –њ—А–Њ–≥—А–µ—Б—Б–Є–≤–љ–Њ –і–µ–≥—А–∞–і–Є—А—Г–µ—В, —В–∞–Ї –Ї–∞–Ї —Б—В—А—Г–Ї—В—Г—А–љ–∞—П –њ–Њ–і–і–µ—А–ґ–Ї–∞ —В–Ї–∞–љ–Є (–≤ —З–∞—Б—В–љ–Њ—Б—В–Є, –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞) —А–∞–Ј—А—Г—И–∞–µ—В—Б—П –±—Л—Б—В—А–µ–µ, —З–µ–Љ —Б–Є–љ—В–µ–Ј–Є—А—Г–µ—В—Б—П.
–Р–љ–∞–ї–Є–Ј –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л–є –≤ –і–∞–љ–љ–Њ–є —Б—В–∞—В—М–µ, –њ–Њ–Ј–≤–Њ–ї–Є–ї —Б—Д–Њ—А–Љ—Г–ї–Є—А–Њ–≤–∞—В—М —А—П–і –њ—А–Є–љ—Ж–Є–њ–Є–∞–ї—М–љ–Њ –љ–Њ–≤—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є –Љ–µ–ґ–і—Г Mg2+ –Є –Ф–°–Ґ. –Э–∞–Є–±–Њ–ї–µ–µ –≤–µ—А–Њ—П—В–љ—Л–Љ–Є –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞–Љ–Є —П–≤–ї—П—О—В—Б—П —Б–ї–µ–і—Г—О—Й–Є–µ: 1) –і–µ—Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є —В–†–Э–Ъ –Є —Б–њ–ї–∞–є—Б–µ–Њ—Б–Њ–Љ; 2) –і–µ–∞–Ї—В–Є–≤–∞—Ж–Є—П –≥–Є–∞–ї—Г—А–Њ–љ–∞–љ—Б–Є–љ—В–µ—В–∞–Ј –Є –њ–Њ–≤—Л—И–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –≥–Є–∞–ї—Г—А–Њ–љ–Є–і–∞–Ј; 3) –∞–Ї—В–Є–≤–∞—Ж–Є—П –Љ–∞—В—А–Є—З–љ—Л—Е –Љ–µ—В–∞–ї–ї–Њ–њ—А–Њ—В–µ–Є–љ–∞–Ј; 4) –Є–љ–∞–Ї—В–Є–≤–∞—Ж–Є—П —Н–ї–∞—Б—В–∞–Ј; 5) –∞–Ї—В–Є–≤–∞—Ж–Є—П —В—А–∞–љ—Б–≥–ї—Г—В–∞–Љ–Є–љ–∞–Ј—Л –Є –ї–Є–Ј–Є–ї–Њ–Ї—Б–Є–і–∞–Ј—Л, –∞ —В–∞–Ї–ґ–µ 6) –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л–µ —А–µ–∞–Ї—Ж–Є–Є, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–µ –∞–ї–ї–µ–ї–µ–Љ bw35 –≥–µ–љ–∞ HLA–B.
–Э–µ–ї—М–Ј—П –Є—Б–Ї–ї—О—З–Є—В—М –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—М —В–Њ–≥–Њ, —З—В–Њ —З–∞—Б—В–Њ –≤—Б—В—А–µ—З–∞—О—Й–Є–µ—Б—П –љ—Г–Ї–ї–µ–Њ—В–Є–і–љ—Л–µ –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ—Л –≤ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–Є—Е –≥–µ–љ–∞—Е –Љ–Њ–≥—Г—В –≤–ї–Є—П—В—М –љ–∞ —Б–Њ—Б—В–Њ—П–љ–Є–µ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Я–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є–µ —Б—Д–Њ—А–Љ—Г–ї–Є—А–Њ–≤–∞–љ–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ –њ–Њ–Ј–≤–Њ–ї–Є—В –њ—А–Њ–≤–µ—Б—В–Є –Њ–±–Њ—Б–љ–Њ–≤–∞–љ–љ—Л–µ –Љ–µ–і–Є–Ї–Њ––≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ—Б—В–≥–µ–љ–Њ–Љ–љ–Њ–≥–Њ —Е–∞—А–∞–Ї—В–µ—А–∞ [79]. –≠–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ—А–µ–і–ї–∞–≥–∞–µ–Љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ –і–Њ–ї–ґ–љ—Л –≤–Ї–ї—О—З–∞—В—М –њ–Њ–і—А–Њ–±–љ—Л–є –∞–љ–∞–ї–Є–Ј —Г–ї—М—В—А–∞—Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –≤ CT, –њ–Њ—А–Њ–ґ–і–∞–µ–Љ—Л—Е Mg2+/Ca2+ –і–µ—Д–Є—Ж–Є—В–Њ–Љ, –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є –Є —Г–ї—М—В—А–∞—Б—В—А—Г–Ї—В—Г—А–љ—Л–є –∞–љ–∞–ї–Є–Ј —В–Ї–∞–љ–µ–є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Ф–°–Ґ, –∞–љ–∞–ї–Є–Ј –≤–Ј–∞–Є–Љ–Њ–Њ—В–љ–Њ—И–µ–љ–Є–є –Љ–µ–ґ–і—Г –∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О –Ь–Ь–Я –Є Mg2+ in vitro, –≤ –Ї–ї–µ—В–Ї–∞—Е –≤ –Ї—Г–ї—М—В—Г—А–µ –∞ —В–∞–Ї–ґ–µ in vivo –Є –і—А.


–Ы–Є—В–µ—А–∞—В—Г—А–∞
1. –§–Є–ї–Є–њ–µ–љ–Ї–Њ –Я.–°., –Ь–∞–ї–Њ–Њ–Ї–∞—П –Ѓ.–°. –†–Њ–ї—М –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –≤ —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–Є –њ—А–Њ–ї–∞–њ—Б–∞ –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞. –Ъ–ї–Є–љ. –Ь–µ–і.,–Ь, 2006;84(12):13–19.
2. –Ъ–ї–µ–Љ–µ–љ–Њ–≤ –Р.–Т. –Э–µ–і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–∞–љ–љ—Л–µ –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ь., 2005. 136 —Б.
3. –Ч–µ–Љ—Ж–Њ–≤—Б–Ї–Є–є –≠.–Т. –Ф–Є—Б–њ–ї–∞—Б—В–Є—З–µ—Б–Ї–Є–µ —Д–µ–љ–Њ—В–Є–њ—Л. –Ф–Є—Б–њ–ї–∞—Б—В–Є—З–µ—Б–Ї–Њ–µ —Б–µ—А–і—Ж–µ. - –°–Я–±.: –Є–Ј–і––≤–Њ «–Ю–ї—М–≥–∞», - 2007. - 80 —Б.
4. –Ь–∞–Ї–Њ–ї–Ї–Є–љ –Т.–Ш., –Я–Њ–і–Ј–Њ–ї–Ї–Њ–≤ –Т.–Ш., –†–Њ–і–Є–Њ–љ–Њ–≤ –Р.–Т., –®–µ—П–љ–Њ–≤ –Ь.–Т., –°–∞–Љ–Њ–є–ї–µ–љ–Ї–Њ –Т.–Т., –Э–∞–њ–∞–ї–Ї–Њ–≤ –Ф.–Р. –†–∞–Ј–љ–Њ–Њ–±—А–∞–Ј–Є–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ґ–µ—А. –Р—А—Е–Є–≤ 2004;76(11):77–80.
5. –Ъ–ї–µ–Љ–µ–љ–Њ–≤ –Р.–Т. –Т–љ–µ–Ї–∞—А–і–Є–∞–ї—М–љ—Л–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –љ–µ–і–Є—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ъ–ї–Є–љ. –Ь–µ–і., –Ь, 2003;81(10):4–7.
6. –Ъ–ї–µ–Љ–µ–љ–Њ–≤ –Р.–Т., –Ь–∞—А—В—Л–љ–Њ–≤ –Т.–Ы., –Ґ–Њ—А–≥—Г—И–Є–љ–∞ –Ш.–°. –Э–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М –±–∞—Г–≥–Є–љ–Є–µ–≤–Њ–є –Ј–∞—Б–ї–Њ–љ–Ї–Є –Ї–∞–Ї –≤–Є—Б—Ж–µp–∞–ї—М–љ–Њ–µ –њp–Њ—П–≤–ї–µ–љ–Є–µ –љ–µ–і–Є—Д—Д–µp–µ–љ—Ж–Єp–Њ–≤–∞–љ–љ–Њ–є –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ґ–µ—А. –Р—А—Е–Є–≤ 2003;75(4):44–46.
7. –С—Г—Е–∞—А–Є–љ –Ю.–Т., –І–µ–њ–∞–ї—М—З–µ–љ–Ї–Њ –Ю.–Х., –Т–∞–ї—Л—И–µ–≤ –Р.–Т., –Х–ї–∞–≥–Є–љ–∞ –Ь.–Ь., –Я–µ—А—Г–љ–Њ–≤–∞ –Э.–С., –Ь–Є—Е–∞–є–ї–Њ–≤–∞ –Х.–Р., –§–Њ–Љ–Є—З–µ–≤–∞ –°.–Т. –Ь–Є–Ї—А–Њ—Д–ї–Њ—А–∞ —В–Њ–ї—Б—В–Њ–≥–Њ –Ї–Є—И–µ—З–љ–Є–Ї–∞ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –і–Є—Б–њ–ї–∞–Ј–Є–µ–є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ц. –Ь–Є–Ї—А–Њ–±–Є–Њ–ї. –≠–њ–Є–і–µ–Љ–Є–Њ–ї. –Ш–Љ–Љ—Г–љ–Њ–±–Є–Њ–ї. 2003;(3):62–66.
8. –Ч–∞—Е–∞—А—П–љ –Р.–Ы., –Ч–∞—Е–∞—А—П–љ –Х.–Р. –Ґ—П–ґ–µ—Б—В—М –≤–∞—А–Є–Ї–Њ–Ј–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є –≤–µ–љ –љ–Є–ґ–љ–Є—Е –Ї–Њ–љ–µ—З–љ–Њ—Б—В–µ–є –њ—А–Є —А–∞–Ј–ї–Є—З–љ—Л—Е —Б—В–µ–њ–µ–љ—П—Е –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ъ–ї–Є–љ. –•–Є—А. 2005;(8):42–44.
9. –¶—Г–Ї–∞–љ–Њ–≤ –Ѓ.–Ґ., –¶—Г–Ї–∞–љ–Њ–≤ –Р.–Ѓ.. –Т–∞—А–Є–Ї–Њ–Ј –љ–Є–ґ–љ–Є—Е –Ї–Њ–љ–µ—З–љ–Њ—Б—В–µ–є –Ї–∞–Ї —А–µ–Ј—Г–ї—М—В–∞—В –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Р–љ–≥–Є–Њ–ї. –°–Њ—Б—Г–і. –•–Є—А. 2004;10(2):84–89.
10. –Ъ–∞–ї–Є–љ–Ї–Є–љ–∞ –Ю.–Ь. –Я—А–Є–Ј–љ–∞–Ї–Є –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –≤ –њ—А–µ–і–њ–Њ–ї–∞–≥–∞–µ–Љ–Њ–є –Є—И–µ–Љ–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є —Б–µ—А–і—Ж–∞. –Ъ–∞—А–і–Є–Њ–ї–Њ–≥–Є—П 1988;28(9):52–57.
11. –Ь–Є—Е–∞–є–ї–Њ–≤–∞ –Р.–Т., –°–Љ–Њ–ї–µ–љ—Б–Ї–Є–є –Р.–Т. –Ю—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ—Л –Є –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є —Д–Є–Ј–Є—З–µ—Б–Ї–Њ–є —А–∞–±–Њ—В–Њ—Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є —Г —Б–њ–Њ—А—В—Б–Љ–µ–љ–Њ–≤ —Б —Б–Є–љ–і—А–Њ–Љ–Њ–Љ –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ъ–ї–Є–љ. –Ь–µ–і., –Ь., 2004;82(8):44–48.
12. –У–Њ–ї–Њ–≤—Б–Ї–Њ–є –С.–Т., –£—Б–Њ–ї—М—Ж–µ–≤–∞ –Ы.–Т., –•–Њ–≤–∞–µ–≤–∞ –ѓ.–Т., –Ш–≤–∞–љ–Њ–≤–∞ –Э.–Т. –Ю—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њp–Њ—П–≤–ї–µ–љ–Є—П –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є —Г –ї–Є—Ж —Вp—Г–і–Њ—Б–њ–Њ—Б–Њ–±–љ–Њ–≥–Њ –≤–Њ–Јp–∞—Б—В–∞. –Ъ–ї–Є–љ. –Ь–µ–і., –Ь, 2002;80(12):39–41.
13. –Ф–Њ–Љ–љ–Є—Ж–Ї–∞—П –Ґ.–Ь., –Ф—К—П—З–µ–љ–Ї–Њ –Р.–Т., –Ъ—Г–њ—А–Є—П–љ–Њ–≤–∞ –Ю.–Ю., –Ф–Њ–Љ–љ–Є—Ж–Ї–Є–є –Ь.–Т. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –њ—А–Є–Љ–µ–љ–µ–љ–Є—П –Љ–∞–≥–љ–Є—П –Њ—А–Њ—В–∞—В–∞ —Г –њ–Њ–і—А–Њ—Б—В–Ї–Њ–≤ —Б —Б–Є–љ–і—А–Њ–Љ–Њ–Љ –і–Є—Б–њ–ї–∞–Ј–Є–Є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є —Б–µ—А–і—Ж–∞. –Ъ–∞—А–і–Є–Њ–ї–Њ–≥–Є—П 2005;45(3):76–81.
14. Alberts B., Johnson A., Lewis J., Raff M., Roberts R., Walter P. Molecular Biology of the Cell, 4th edition, Garland Science, 2002, ISBN 0815340729.
15. Gleghorn L., Ramesar R., Beighton P., Wallis G. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am J Hum Genet. 2005;77(3):484–90 Epub 2005 Ju.
16. Nicole S., Davoine C.S., Topaloglu H., Cattolico L., Barral D., Beighton P., Hamida C.B., Hammouda H., Cruaud C., White P.S., Samson D., Urtizberea J.A., Lehmann–Horn F., Weissenbach J., Hentati F., Fontaine B. Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz–Jampel syndrome (chondrodystrophic myotonia). Nat Genet. 2000;26(4):480–483.
17. Jepsen K.J., Wu F., Peragallo J.H., Paul J., Roberts L., Ezura Y., Oldberg A., Birk D.E., Chakravarti S. A syndrome of joint laxity and impaired tendon integrity in lumican– and fibromodulin–deficient mice. J Biol Chem. 2002;277(38):35532–40 Epub 2002.
18. Okajima T., Fukumoto S., Furukawa K., Urano T. Molecular basis for the progeroid variant of Ehlers–Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J Biol Chem. 1999;274(41):28841–28844.
19. Zweers M.C., Dean W.B., van Kuppevelt T.H., Bristow J., Schalkwijk J. Elastic fiber abnormalities in hypermobility type Ehlers–Danlos syndrome patients with tenascin–X mutations. Clin Genet. 2005;67(4):330–334.
20. Cabral W.A., Chang W., Barnes A.M., Weis M., Scott M.A., Leikin S., Makareeva E., Kuznetsova N.V., Rosenbaum K.N., Tifft C.J., Bulas D.I., Kozma C., Smith P.A., Eyre D.R., Marini J.C. Prolyl 3–hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007;39(3):359–65 Epub 2007 Fe.
21. Yeowell H.N., Walker L.C. Ehlers–Danlos syndrome type VI results from a nonsense mutation and a splice site–mediated exon–skipping mutation in the lysyl hydroxylase gene. Proc Assoc Am Physicians. 1997;109(4):383–396.
22. van der Slot A.J., Zuurmond A.M., Bardoel A.F., Wijmenga C., Pruijs H.E., Sillence D.O., Brinckmann J., Abraham D.J., Black C.M., Verzijl N., DeGroot J., Hanemaaijer R., TeKoppele J.M., Huizinga T.W., Bank R.A. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J Biol Chem. 2003;278(42):40967–72 Epub 2003.
23. Colige A., Sieron A.L., Li S.W., Schwarze U., Petty E., Wertelecki W., Wilcox W., Krakow D., Cohn D.H., Reardon W., Byers P.H., Lapiere C.M., Prockop D.J., Nusgens B.V. Human Ehlers–Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N–proteinase gene. Am J Hum Genet. 1999;65(2):308–317.
24. Di Ferrante N., Leachman R.D., Angelini P., Donnelly P.V., Francis G., Almazan A. Lysyl oxidase deficiency in Ehlers–Danlos syndrome type V. Connect Tissue Res. 1975;3(1):49–53.
25. Cauwe B., Van den Steen P.E., Opdenakker G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit Rev Biochem Mol Biol. 2007;42(3):113–185.
26. Malemud C.J. Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci. 2006;11:1696–1701.
27. Shingleton W.D., Hodges D.J., Brick P., Cawston T.E. Collagenase: a key enzyme in collagen turnover. Biochem Cell Biol. 1996;74(6):759–775.
28. Elastin: mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet. 2000;8(12):955–963.
29. Zacchigna L., Vecchione C., Notte A., Cordenonsi M., et al. Emilin1 links TGF–beta maturation to blood pressure homeostasis. Cell. 2006;124(5):929–942.
30. Loeys B.L., Schwarze U., Holm T., Callewaert B.L., Thomas G.H., Pannu H., De Backer J.F. et al. Aneurysm syndromes caused by mutations in the TGF–beta receptor. N Engl J Med. 2006;355(8):788–798.
31. –°–њ–∞—Б–Њ–≤ –Р.–Р. –Ь–∞–≥–љ–Є–є –≤ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–є –њ—А–∞–Ї—В–Є–Ї–µ. - –Т–Њ–ї–≥–Њ–≥—А–∞–і, 2000. – 272 —Б.
32. Senni K., Foucault–Bertaud A., Godeau G. Magnesium and connective tissue. Magnes Res. 2003;16(1):70–74.
33. –У—А–Њ–Љ–Њ–≤–∞ –Ю.–Р. –Ь–∞–≥–љ–Є–є –Є –њ–Є—А–Є–і–Њ–Ї—Б–Є–љ: –Њ—Б–љ–Њ–≤—Л –Ј–љ–∞–љ–Є–є. - –Ь.: –Я—А–Њ—В–Њ–Ґ–Є–њ, 2006. – 234 —Б.
34. Quamme G.A., de Rouffignac C. Epithelial magnesium transport and regulation by the kidney. Front Biosci. 2000;5:D694–D711.
35. Konrad M., Weber S. Recent advances in molecular genetics of hereditary magnesium–losing disorders. J Am Soc Nephrol. 2003;14(1):249–260.
36. –У—А–Њ–Љ–Њ–≤–∞ –Ю.–Р, –У–Њ–≥–Њ–ї–µ–≤–∞ –Ш.–Т. –Я—А–Є–Љ–µ–љ–µ–љ–Є–µ –Љ–∞–≥–љ–Є—П –≤ –Ј–µ—А–Ї–∞–ї–µ –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М–љ–Њ–є –Љ–µ–і–Є—Ж–Є–љ—Л –Є —Д—Г–љ–і–∞–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –≤ —В–µ—А–∞–њ–Є–Є //–§–∞—А–Љ–∞—В–µ–Ї–∞. - 2007. - —В–Њ–Љ 146, вДЦ12. - –°. 3–6].
37. Chubanov V., Waldegger S., Mederos y Schnitzler M., Vitzthum H., Sassen M.C., Seyberth H.W., Konrad M., Gudermann T. Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci U S A. 2004;101(9):2894–9.
38. Schlingmann K.P., Weber S., Peters M., Niemann Nejsum L., Vitzthum H., Klingel K., Kratz M., Haddad E., Ristoff E., Dinour D., Syrrou M., Nielsen S., Sassen M., Waldegger S., Seyberth H.W., Konrad M. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet. 2002;31(2):166–70.
39. Wang Z., Hu S.Y., Lei D.L., Song W.X. Effect of chronic stress on PKA and P–CREB expression in hippocampus of rats and the antagonism of antidepressors Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2006;31(5):767–771.
40. Goytain A., Quamme G.A. Functional characterization of human SLC41A1, a Mg2+ transporter with similarity to prokaryotic MgtE Mg2+ transporters. Physiol Genomics. 2005;21(3):337–42 Epub 2005 Fe.
41. Delprat B., Bibert S., Geering K. FXYD proteins: novel regulators of Na,K–ATPase Med Sci (Paris). 2006;22(6–7):633–638.
42. Meij I.C., Koenderink J.B., van Bokhoven H., Assink K.F., Groenestege W.T., de Pont JJ.., Bindels R.J., Monnens L.A., van den Heuvel L.P., Knoers N.V. Dominant isolated renal magnesium loss is caused by misrouting of the Na(+),K(+)–ATPase gamma–subunit. Nat Genet. 2000;26(3):265–266.
43. Lee NP, Tong MK, Leung PP, Chan VW, Leung S, Tam PC, Chan KW, Lee KF, Yeung WS, Luk JM. Kidney claudin–19: localization in distal tubules and collecting ducts and dysregulation in polycystic renal disease. FEBS Lett. 2006;580(3):923–31 Epub 2006 Ja.
44. Simon DB, Lu Y, Choate KA, Velazquez H, Al–Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez–Soriano J, McCredie D, Milford D, Sanjad S, Lifton RP. Paracellin–1, a renal tight junction protein required for paracellular Mg2+ resorption. Science. 1999;285(5424):103–106.
45. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, Vitzthum H, Suzuki Y, et al. Mutations in the tight–junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79(5):949–57.
46. Nagase T, Murakami T, Tsukada T, Kitamura R, Chikatsu N, Takeo H, Takata N, Yasuda H, Fukumoto S, Tanaka Y, Nagata N, Yamaguchi K, Akatsu T, Yamamoto M. A family of autosomal dominant hypocalcemia with a positive correlation between serum calcium and magnesium: identification of a novel gain of function mutation (Ser(820)Phe) in the calcium–sensing receptor. J Clin Endocrinol Metab. 2002;87(6):2681–2687.
47. Hendy GN, D’Souza–Li L, Yang B, Canaff L, Cole DE. Mutations of the calcium–sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat. 2000;16(4):281–296.
48. Ikari A, Okude C, Sawada H, Sasaki Y, Yamazaki Y, Sugatani J, Degawa M, Miwa M. Activation of a polyvalent cation–sensing receptor decreases magnesium transport via claudin–16. Biochim Biophys Acta. 2007;
49. Liang Z, Yang Z. Identification and characterization of a novel gene EOLA1 stimulating ECV304 cell proliferation. Biochem Biophys Res Commun. 2004;325(3):798–802.
50. Feng W, Benz FW, Cai J, Pierce WM, Kang YJ. Metallothionein disulfides are present in metallothionein–overexpressing transgenic mouse heart and increase under conditions of oxidative stress. J Biol Chem. 2006;281(2):681–7.
51. Giacconi R, Muti E, Malavolta M, Cipriano C, Costarelli L, Bernardini G, Gasparini N, Mariani E, Saba V, Boccoli G, Mocchegiani E. The +838 C/G MT2A polymorphism, metals, and the inflammatory/immune response in carotid artery stenosis in elderly people. Mol Med. 2007;13(7–8):388–395.
52. Laurant P, Hayoz D, Brunner H, Berthelot A. Dietary magnesium intake can affect mechanical properties of rat carotid artery. Br J Nutr. 2000;84(5):757–764.
53. Leonard F, Moscovitz P, Hodge JW, Adams JP. Age related Ca–Mg content and strength in turkey tendon. Calcif Tissue Res. 1976;19(4):331–336.
54. –Ъ–∞–њ–µ–ї—М–Ї–Њ –Т.–Ш. –Т–љ–µ–Ї–ї–µ—В–Њ—З–љ—Л–є –Љ–∞—В—А–Є–Ї—Б –Љ–Є–Њ–Ї–∞—А–і–∞ –Є –µ–≥–Њ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ—А–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е —Б–µ—А–і—Ж–∞ //–Ъ–∞—А–і–Є–Њ–ї–Њ–≥–Є—П. – 2000. – вДЦ9. – –°. 78–90.)
55. van Venrooij WJ. Autoantibodies against small nuclear ribonucleoprotein components. J Rheumatol Suppl. 1987;14 Suppl 1:78–82.
56. Mio K, Carrette O, Maibach HI, Stern R. Evidence that the serum inhibitor of hyaluronidase may be a member of the inter–alpha–inhibitor family. J Biol Chem. 2000;275(42):32413–32421.
57. Ueshima K, Shibata M, Suzuki T, Endo S, Hiramori K. Extracellular matrix disturbances in acute myocardial infarction: relation between disease severity and matrix metalloproteinase–1, and effects of magnesium pretreatment on reperfusion injury. Magnes Res. 2003;16(2):120–126.
58. Guo H, Lee JD, Uzui H, Yue H, Wang J, Toyoda K, Geshi T, Ueda T. Effects of folic acid and magnesium on the production of homocysteine–induced extracellular matrix metalloproteinase–2 in cultured rat vascular smooth muscle cells. Circ J. 2006;70(1):141–146.
59. Pages N, Gogly B, Godeau G, Igondjo–Tchen S, Maurois P, Durlach J, Bac P. Structural alterations of the vascular wall in magnesium–deficient mice. A possible role of gelatinases A (MMP–2) and B (MMP–9). Magnes Res. 2003;16(1):43–48.
60. Yue H, Lee JD, Shimizu H, Uzui H, Mitsuke Y, Ueda T. Effects of magnesium on the production of extracellular matrix metalloproteinases in cultured rat vascular smooth muscle cells. Atherosclerosis. 2003;166(2):271–277.
61. Rabaud M, Lefebvre F, Graves PV, Desgranges C, Bricaud H. Magnesium enhances human pancreatic elastase digestion of 125I–labeled elastin. Experientia. 1985;41(5):628–631.
62. Cardy CM, Handford PA. Metal ion dependency of microfibrils supports a rod–like conformation for fibrillin–1 calcium–binding epidermal growth factor–like domains. J Mol Biol. 1998;276(5):855–860.
63. Clarke AW, Weiss AS. Microfibril–associated glycoprotein–1 binding to tropoelastin: multiple binding sites and the role of divalent cations. Eur J Biochem. 2004;271(14):3085–3090.
64. Ahvazi B, Boeshans KM, Rastinejad F. The emerging structural understanding of transglutaminase 3. J Struct Biol. 2004;147(2):200–207.
65. Ahvazi B, Boeshans KM, Idler W, Baxa U, Steinert PM. Roles of calcium ions in the activation and activity of the transglutaminase 3 enzyme. J Biol Chem. 2003;278(26):23834–41 Epub 2003.
66. Gacheru SN, Trackman PC, Shah MA, O’Gara CY, Spacciapoli P, Greenaway FT, Kagan HM. Structural and catalytic properties of copper in lysyl oxidase. J Biol Chem. 1990;265(31):19022–19027.
67. Kachru RB, Telischi M, Cruz JB, Patel R, Towne WD. The HLA antigens and ABO blood groups in an American Black population with mitral valve prolapse. Tissue Antigens. 1979;14(3):256–260.
68. Zeana CD. Recent data on mitral valve prolapse and magnesium deficit. Magnes Res. 1988;1(3–4):203–211.
69. Henrotte JG. The variability of human red blood cell magnesium level according to HLA groups. Tissue Antigens. 1980;15(5):419–430.
70. Durlach J. Recommended dietary amounts of magnesium: Mg RDA. Magnes Res. 1989;2(3):195–203.
71. Miceli–Richard C, Zouali H, Said–Nahal R, Lesage S, Merlin F, De Toma C, Blanche H, Sahbatou M, Dougados M, Thomas G, Breban M, Hugot JP. Significant linkage to spondyloarthropathy on 9q31–34. Hum Mol Genet. 2004;13(15):1641–1648.
72. Rubin LA, Amos CI, Wade JA, Martin JR, Bale SJ, Little AH, Gladman DD, Bonney GE, Rubenstein JD, Siminovitch KA. Investigating the genetic basis for ankylosing spondylitis. Linkage studies with the major histocompatibility complex region. Arthritis Rheum. 1994;37(8):1212–1220.
73. Forsberg B, Low B. Malignant hypertension and HLA antigens. Tissue Antigens. 1983;22(2):155–159.
74. Laurentaci G, Lomuto M, Favoino B. Immunogenetic analysis of association between HLA antigens and psoriasis vulgaris: population and family studies. Dermatologica. 1982;165(6):591–600.
75. Walker WG, Hillis WD, Hillis A. Hepatitis B infection in patients with end stage renal disease: some characteristics and consequences. Trans Am Clin Climatol Assoc. 1980;92:142–151.
76. Ludwig H, Mayr W. Genetic aspects of susceptibility to multiple myeloma. Blood. 1982;59(6):1286–1291.
77. Nyulassy S, Hnilica P, Buc M, Guman M, Hirschova V, Stefanovic J. Subacute (de Quervain’s) thyroiditis: association with HLA–Bw35 antigen and abnormalities of the complement system, immunoglobulins and other serum proteins. J Clin Endocrinol Metab. 1977;45(2):270–274.
78. Mehra NK, Ahuja GK, Taneja V, Vaidya MC. HLA antigens and myasthenia gravis in north India. J Neurol Neurosurg Psychiatry. 1983;46(4):361–364.
79. Torshin I.Yu. Bioinformatics in the post–genomic era: physiology and medicine. Nova Biomedical Books, NY, USA (2007), ISBN: 1600217524, pp35–67.



