




–Я—А–Њ—Д–µ—Б—Б–Њ—А –Р.–Р. –Р–ї–µ–Ї—Б–∞–љ–і—А–Њ–≤
–≠–љ–і–Њ–Ї—А–Є–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є –Э–∞—Г—З–љ—Л–є –¶–µ–љ—В—А –†–Р–Ь–Э, –Ь–Њ—Б–Ї–≤–∞–Ъ–Њ–љ—В—А–Њ–ї—М –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П, –њ–Њ–Ј–≤–Њ–ї—П—О—Й–Є–є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ —Г–ї—Г—З—И–∞—В—М –њ—А–Њ–≥–љ–Њ–Ј –Ш–С–° —Г –±–Њ–ї—М–љ—Л—Е —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ 2 —В–Є–њ–∞, –Ј–∞–љ–Є–Љ–∞–µ—В —Б–µ–є—З–∞—Б –њ—А–Є–Њ—А–Є—В–µ—В–љ–Њ–µ, –њ–Њ —Б—Г—В–Є –і–µ–ї–∞, –њ–µ—А–≤–Њ–µ –Љ–µ—Б—В–Њ –Љ–µ–ґ–і—Г –і—А—Г–≥–Є–Љ–Є –∞–љ–∞–ї–Њ–≥–Є—З–љ—Л–Љ–Є –≤–Љ–µ—И–∞—В–µ–ї—М—Б—В–≤–∞–Љ–Є. –Ъ —В–∞–Ї–Њ–Љ—Г –≤—Л–≤–Њ–і—Г –њ—А–Є—И–ї–Є –∞–≤—В–Њ—А—Л United Kingdom Prospective Diabetes Study, —Е–Њ—А–Њ—И–Њ –Є–Ј–≤–µ—Б—В–љ–Њ–≥–Њ –Є —В—Й–∞—В–µ–ї—М–љ–Њ –њ—А–Њ–і—Г–Љ–∞–љ–љ–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –њ–Њ—Б–≤—П—Й–µ–љ–љ–Њ–≥–Њ –Є–Ј—Г—З–µ–љ–Є—О —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є —Г –±–Њ–ї—М–љ—Л—Е —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ 2 —В–Є–њ–∞ (UKPDS 38, 1998). –Ш–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –Ї–Њ–љ—В—А–Њ–ї—М —Г—А–Њ–≤–љ—П –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П –њ–Њ–Ј–≤–Њ–ї—П–µ—В —Б–љ–Є–Ј–Є—В—М —А–∞–Ј–≤–Є—В–Є–µ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є —Г –±–Њ–ї—М–љ—Л—Е —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ 2 —В–Є–њ–∞ –љ–∞ 51%, –≤ —В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї –Ї–Њ–љ—В—А–Њ–ї—М –≥–Є–њ–µ—А–ї–Є–њ–Є–і–µ–Љ–Є–Є —Б–љ–Є–ґ–∞–µ—В –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є –Њ—В –Ш–С–° –љ–∞ 36%, –∞ —В—Й–∞—В–µ–ї—М–љ–∞—П –Ї–Њ—А—А–µ–Ї—Ж–Є—П —Г—А–Њ–≤–љ—П –≥–ї–Є–Ї–µ–Љ–Є–Є —Б–љ–Є–ґ–∞–µ—В —З–∞—Б—В–Њ—В—Г —А–∞–Ј–≤–Є—В–Є—П –Є–љ—Д–∞—А–Ї—В–Њ–≤ –Љ–Є–Њ–Ї–∞—А–і–∞ –љ–∞ 16% (IDF, 1999).
–Ъ–∞–Ї —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В —А–µ–Ј—Г–ї—М—В–∞—В—Л –љ–∞—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е –њ—А–Њ—Д–Є–ї–∞–Ї—В–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ–≥—А–∞–Љ–Љ —А–∞–Ј–ї–Є—З–љ—Л—Е —Б—В—А–∞–љ, –љ–Њ—А–Љ–∞–ї–Є–Ј–∞—Ж–Є—П –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П, –Ї—А–Њ–Љ–µ —В–Њ–≥–Њ, –Є–≥—А–∞–µ—В –≤–µ–і—Г—Й—Г—О —А–Њ–ї—М –≤ –њ—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–µ —Б–∞—Е–∞—А–љ–Њ–≥–Њ –і–Є–∞–±–µ—В–∞ —Г –ї–Є—Ж —Б –≤—Л—Б–Њ–Ї–Є–Љ —А–Є—Б–Ї–Њ–Љ –µ–≥–Њ —А–∞–Ј–≤–Є—В–Є—П (–Ъ–Њ–њ–µ–љ–≥–∞–≥–µ–љ, 1996).
–°–Њ—З–µ—В–∞–љ–Є–µ –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–Є —Б –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–µ–є —Е–Њ—А–Њ—И–Њ –Є–Ј–≤–µ—Б—В–љ–Њ. –Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –Њ–љ–Њ –≤—Б—В—А–µ—З–∞–µ—В—Б—П —Г –±–Њ–ї—М–љ—Л—Е —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ 2 —В–Є–њ–∞ –Є —Г –ї–Є—Ж —Б –Њ–ґ–Є—А–µ–љ–Є–µ–Љ (Christlieb A.R. et al., 1985; Manicardi V. et al., 1986; Rose H. et al., 1987; Modan M., Halkin H., Almog S. et al., 1985; Tappy et al.,1991; Fournier A.M. et al., 1986).
–°–∞–Љ –њ–Њ —Б–µ–±–µ –Є–љ—Б—Г–ї–Є–љ –Њ–±–ї–∞–і–∞–µ—В –њ—А—П–Љ—Л–Љ —Б–Њ—Б—Г–і–Њ—А–∞—Б—И–Є—А—П—О—Й–Є–Љ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ–Љ (Anderson E.A., Mark A.L.,1993). –£ –љ–Њ—А–Љ–∞–ї—М–љ—Л—Е –ї—О–і–µ–є –≤–≤–µ–і–µ–љ–Є–µ —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –і–Њ–Ј –Є–љ—Б—Г–ї–Є–љ–∞ –њ—А–Є –Њ—В—Б—Г—В—Б—В–≤–Є–Є –≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є–Є –≤—Л–Ј—Л–≤–∞–µ—В –≤–∞–Ј–Њ–і–Є–ї–∞—В–∞—Ж–Є—О, –∞ –љ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П (Anderson E.A., Mark A.L.,1993; Anderson E.A. et al., 1991). –°—А–µ–і–Є –±–Њ–ї—М–љ—Л—Е —Б –Є–љ—Б—Г–ї–Є–љ–Њ–Љ–∞–Љ–Є –∞—А—В–µ—А–Є–∞–ї—М–љ–∞—П –≥–Є–њ–µ—А—В–Њ–љ–Є—П –≤—Б—В—А–µ—З–∞–µ—В—Б—П –љ–µ —З–∞—Й–µ, —З–µ–Љ —Г –ї–Є—Ж –±–µ–Ј –Є–љ—Б—Г–ї–Є–љ–Њ–Љ (Sawicki P.T. et al., 1992).
–Ш–Љ–µ—О—В—Б—П —Г–±–µ–і–Є—В–µ–ї—М–љ—Л–µ –і–∞–љ–љ—Л–µ –Њ —В–Њ–Љ, —З—В–Њ –≤–Є–і–Њ–≤—Л–µ –Є —А–∞—Б–Њ–≤—Л–µ –Њ—В–ї–Є—З–Є—П –≤–Њ –Љ–љ–Њ–≥–Њ–Љ –Њ–њ—А–µ–і–µ–ї—П—О—В —А–µ–∞–Ї—Ж–Є—О –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П –љ–∞ –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є—О (Landsberg L., 1986). –Я–Њ –і–∞–љ–љ—Л–Љ —Н–њ–Є–і–µ–Љ–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, —Г –±–µ–ї—Л—Е –ї–Є—Ж –±–µ–Ј –≥–Є–њ–µ—А—В–Њ–љ–Є–Є —Г—А–Њ–≤–µ–љ—М –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ –Ї–Њ—А—А–µ–ї–Є—А—Г–µ—В —Б –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–µ–є –Є–љ—Б—Г–ї–Є–љ–∞ –≤ –њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є –љ–∞—В–Њ—Й–∞–Ї, –≤ —В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї —Г –∞—Д—А–Њ––∞–Љ–µ—А–Є–Ї–∞–љ—Ж–µ–≤ –Є —Г –Є–љ–і–µ–є—Ж–µ–≤ –Я–Є–Љ–∞ —В–∞–Ї–Є—Е –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–µ–є –љ–µ –Њ—В–Љ–µ—З–µ–љ–Њ (Saad M.F. et al., 1991). C –і—А—Г–≥–Њ–є —Б—В–Њ—А–Њ–љ—Л, –і–ї—П –∞—Д—А–Њ––∞–Љ–µ—А–Є–Ї–∞–љ—Ж–µ–≤ —Б –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–µ–є –≤ –Њ—В–ї–Є—З–Є–µ –Њ—В –∞—Д—А–Њ––∞–Љ–µ—А–Є–Ї–∞–љ—Ж–µ–≤ –±–µ–Ј –≥–Є–њ–µ—А—В–Њ–љ–Є–Є —Е–∞—А–∞–Ї—В–µ—А–љ–Њ –љ–∞–ї–Є—З–Є–µ –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–Є –Є –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є, —З—В–Њ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –Њ —Б–≤—П–Ј–Є –Љ–µ–ґ–і—Г –њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П –Є —А–Њ—Б—В–Њ–Љ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Є–љ—Б—Г–ї–Є–љ–∞ –≤ –њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є —Г —Н—В–Њ–є –≥—А—Г–њ–њ—Л –ї–Є—Ж (Falkner B., Hulman S., Kushner H., 1993).
–Ю—В–Љ–µ—З–µ–љ–Њ, —З—В–Њ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–µ –і–∞–≤–ї–µ–љ–Є–µ —Г–Љ–µ–љ—М—И–∞–µ—В—Б—П –њ—А–Є —Б–љ–Є–ґ–µ–љ–Є–Є –і–Њ–Ј—Л –Є–љ—Б—Г–ї–Є–љ–∞ —Г –±–Њ–ї—М–љ—Л—Е —Б –њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ –≤–µ—Б–Њ–Љ, —Б—В—А–∞–і–∞—О—Й–Є—Е —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ 2 —В–Є–њ–∞ (Tedde R. et al, 1989). –Ш–Ј–≤–µ—Б—В–љ–Њ —В–∞–Ї–ґ–µ, —З—В–Њ –њ—А–Є –љ–∞—З–∞–ї–µ –Є–љ—Б—Г–ї–Є–љ–Њ—В–µ—А–∞–њ–Є–Є —Г –ї–Є—Ж —Б –њ–ї–Њ—Е–Њ –Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ—Л–Љ —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ 2 —В–Є–њ–∞ —Г—А–Њ–≤–µ–љ—М –Є—Е –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П –љ–∞—А–∞—Б—В–∞–µ—В (Randeree H.A. et al, 1992).
–†–µ–∞–Ї—Ж–Є—П –љ–∞ –≤–≤–µ–і–µ–љ–Є–µ –Є–љ—Б—Г–ї–Є–љ–∞ —Г —З–µ–ї–Њ–≤–µ–Ї–∞ —Б–≤–Њ–є—Б—В–≤–µ–љ–љ–∞ –Є –љ–µ–Ї–Њ—В–Њ—А—Л–Љ –ґ–Є–≤–Њ—В–љ—Л–Љ. –Ъ—А–∞—В–Ї–Њ–≤—А–µ–Љ–µ–љ–љ—Л–µ –Є –і–ї–Є—В–µ–ї—М–љ—Л–µ –Є–љ—Д—Г–Ј–Є–Є –Є–љ—Б—Г–ї–Є–љ–∞ —Б–Њ–±–∞–Ї–∞–Љ –љ–µ –њ–Њ–≤—Л—И–∞—О—В —Г –љ–Є—Е –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П (Hall J.E. et al., 1990; Hall J.E., Colman T.G., Mizelle H.L., 1989). –Т —В–Њ –ґ–µ –≤—А–µ–Љ—П –≤–≤–µ–і–µ–љ–Є–µ –Є–љ—Б—Г–ї–Є–љ–∞ –Ї—А—Л—Б–∞–Љ –≤–µ–і–µ—В –Ї –њ–Њ–≤—Л—И–µ–љ–Є—О –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П (Brands M.W. et al., 1991; Hsieh P.S., Huang W.C., 1993; Tomiyama H. et al., 1992; Meehan W.P. et al., 1994). –£ –Ї—А—Л—Б –њ–Њ–≤—Л—И–µ–љ–Є–µ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П, –≤—Л–Ј–≤–∞–љ–љ–Њ–µ –≤–≤–µ–і–µ–љ–Є–µ–Љ –Є–љ—Б—Г–ї–Є–љ–∞, –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Г–Љ–µ–љ—М—И–∞–µ—В—Б—П –њ—А–Є –њ—А–µ–Ї—А–∞—Й–µ–љ–Є–Є –Є–љ—Б—Г–ї–Є–љ–Њ–≤—Л—Е –Є–љ—Д—Г–Ј–Є–є, –њ–Њ–і—В–≤–µ—А–ґ–і–∞—П —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–µ –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Њ–љ–љ–Њ–µ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–Є (Hsieh P.S., Huang W.C., 1993).
–£—З–Є—В—Л–≤–∞—П —В–Њ, —З—В–Њ –Є–љ—Б—Г–ї–Є–љ —П–≤–ї—П–µ—В—Б—П –њ—А—П–Љ—Л–Љ –≤–∞–Ј–Њ–і–Є–ї–∞—В–∞—В–Њ—А–Њ–Љ, —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є –њ—А–Є –µ–≥–Њ —Г—З–∞—Б—В–Є–Є, –њ–Њ––≤–Є–і–Є–Љ–Њ–Љ—Г, –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –≤–Њ –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є–Є —Б –і—А—Г–≥–Є–Љ–Є —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞–Љ–Є. –Я—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ —Н—В–Њ –Њ—В–љ–Њ—Б–Є—В—Б—П –Ї –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є—О –Є–љ—Б—Г–ї–Є–љ–∞ –Є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л.
–Т —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л—Е —Г—Б–ї–Њ–≤–Є—П—Е —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ –њ–Є—Й–Є –њ–Њ–≤—Л—И–∞–µ—В, –∞ –≥–Њ–ї–Њ–і–∞–љ–Є–µ —Б–љ–Є–ґ–∞–µ—В –∞–Ї—В–Є–≤–љ–Њ—Б—В—М —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л (Young J.B., Landsberg L., 1977). –Я–Њ–і–Њ–±–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤ —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ–Њ–і –≤–ї–Є—П–љ–Є–µ–Љ –њ–Є—Й–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –Є —Г –ї—О–і–µ–є (Landsberg L., Young J.B., 1985). –Т –њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М —Н—В–Њ –≤—Л—П–≤–ї—П–µ—В—Б—П –њ—А–Є —Г–≤–µ–ї–Є—З–µ–љ–Є–Є –њ–Њ—В—А–µ–±–ї–µ–љ–Є—П —Г–≥–ї–µ–≤–Њ–і–Њ–≤ –Є –ґ–Є—А–Њ–≤ (Young J.B. et al., 1982; Schwartz J.H., Young J.B., Landsberg L., 1983; Kaufman L.N., Young J.B., Landsberg l., 1986).
–Ъ–∞–Ї –Њ–Ї–∞–Ј–∞–ї–Њ—Б—М, –Є–љ—Б—Г–ї–Є–љ –Є–≥—А–∞–µ—В –Ї–ї—О—З–µ–≤—Г—О —А–Њ–ї—М –≤–Њ –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є –њ–Њ—В—А–µ–±–ї–µ–љ–Є—П –њ–Є—Й–Є –Є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Є––Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ–Њ–є —В—А–∞—В–Њ–є —Н–љ–µ—А–≥–Є–Є (Landsberg L., Young J.B., 1985). –Я–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞ –њ–Є—Й–Є —Б–µ–Ї—А–µ—Ж–Є—П –Є–љ—Б—Г–ї–Є–љ–∞ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П. –Я—А–Є —Н—В–Њ–Љ –Є–љ—Б—Г–ї–Є–љ —Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В –њ–Њ—В—А–µ–±–ї–µ–љ–Є–µ –Є –Њ–±–Љ–µ–љ –≥–ї—О–Ї–Њ–Ј—Л –≤ —А–µ–≥—Г–ї—П—В–Њ—А–љ—Л—Е –Ї–ї–µ—В–Ї–∞—Е, –∞–љ–∞—В–Њ–Љ–Є—З–µ—Б–Ї–Є —Б–≤—П–Ј–∞–љ–љ—Л—Е —Б –≤–µ–љ—В—А–Њ–Љ–µ–і–Є–∞–ї—М–љ—Л–Љ–Є —П–і—А–∞–Љ–Є –≥–Є–њ–Њ—В–∞–ї–∞–Љ—Г—Б–∞ (Landsberg L., Young J.B., 1985). –£–≤–µ–ї–Є—З–µ–љ–Є–µ –њ–Њ—В—А–µ–±–ї–µ–љ–Є—П –≥–ї—О–Ї–Њ–Ј—Л –≤ —Н—В–Є—Е –љ–µ–є—А–Њ–љ–∞—Е –≤–µ–і–µ—В –Ї —Г–Љ–µ–љ—М—И–µ–љ–Є—О –Є—Е —Г–≥–љ–µ—В–∞—О—Й–µ–≥–Њ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –љ–∞ —Б—В–≤–Њ–ї –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –Т —А–µ–Ј—Г–ї—М—В–∞—В–µ —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ—Л–µ —В–∞–Љ —Ж–µ–љ—В—А—Л —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є —А–µ–≥—Г–ї—П—Ж–Є–Є —А–∞—Б—В–Њ—А–Љ–∞–ґ–Є–≤–∞—О—В—Б—П –Є —Ж–µ–љ—В—А–∞–ї—М–љ–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л (–°–Э–°) –љ–∞—А–∞—Б—В–∞–µ—В (Landsberg L., Young J., 1985; Gans R.O., Donker A.J., 1991) (—А–Є—Б. 1). –Я–Њ–≤—Л—И–µ–љ–Є–µ —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞ –њ–Є—Й–Є –≤–µ–і–µ—В –Ї –љ–∞—А–∞—Б—В–∞–љ–Є—О –њ–Њ—В—А–µ–±–ї–µ–љ–Є—П —Н–љ–µ—А–≥–µ—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–њ–∞—Б–Њ–≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–∞. –Ь–µ—Е–∞–љ–Є–Ј–Љ –њ–Є—Й–µ–≤–Њ–є —А–µ–≥—Г–ї—П—Ж–Є–Є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –°–Э–° –њ–Њ–Ј–≤–Њ–ї—П–µ—В —Н–Ї–Њ–љ–Њ–Љ–Є—В—М —А–∞—Б—Е–Њ–і –Ї–∞–ї–Њ—А–Є–є –≤ –њ–µ—А–Є–Њ–і –≥–Њ–ї–Њ–і–∞–љ–Є—П –Є —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —Б–ґ–Є–≥–∞–љ–Є—О –Є–Ј–±—Л—В–Њ—З–љ—Л—Е –Ї–∞–ї–Њ—А–Є–є –њ—А–Є –њ–µ—А–µ–µ–і–∞–љ–Є–Є. –Х–µ —Н—Д—Д–µ–Ї—В –љ–∞–њ—А–∞–≤–ї–µ–љ –љ–∞ —Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є—О —Н–љ–µ—А–≥–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –±–∞–ї–∞–љ—Б–∞ –Њ—А–≥–∞–љ–Є–Ј–Љ–∞ –Є —Б–Њ—Е—А–∞–љ–µ–љ–Є–µ —Г—Б—В–Њ–є—З–Є–≤–Њ–≥–Њ –≤–µ—Б–∞ —В–µ–ї–∞. –Ъ–ї—О—З–µ–≤–∞—П —А–Њ–ї—М –Є–љ—Б—Г–ї–Є–љ–∞ –≤ —А–µ–∞–ї–Є–Ј–∞—Ж–Є–Є –і–µ–є—Б—В–≤–Є—П —Н—В–Њ–≥–Њ –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞ –≤–њ–Њ–ї–љ–µ –Њ—З–µ–≤–Є–і–љ–∞ (Young J.B., Landsberg L., 1984).
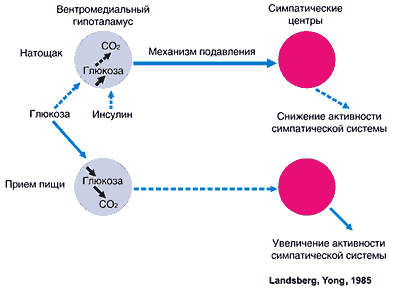
–†–Є—Б. 1. –Ш–љ—Б—Г–ї–Є–љ –Є –≥–ї—О–Ї–Њ–Ј–∞ –≤ —А–µ–≥—Г–ї—П—Ж–Є–Є —Ж–µ–љ—В—А–∞–ї—М–љ–Њ–є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є
–Я—А–Є –Њ–ґ–Є—А–µ–љ–Є–Є –Ї–Њ–Љ–њ–µ–љ—Б–∞—В–Њ—А–љ–∞—П –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є—П, –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–∞—П –љ–∞ —Д–Њ–љ–µ —Б–љ–Є–ґ–µ–љ–Є—П —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є –Ї –Є–љ—Б—Г–ї–Є–љ—Г, –≤–µ–і–µ—В –Ї –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–Љ—Г —А–Њ—Б—В—Г –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —Ж–µ–љ—В—А–∞–ї—М–љ—Л—Е —П–і–µ—А –°–Э–°. –Ш –≤ —Н—В–Њ–Љ —Б–ї—Г—З–∞–µ –њ–Њ–≤—Л—И–µ–љ–љ–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –°–Э–° —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ –љ–∞–њ—А–∞–≤–ї–µ–љ–∞ –љ–∞ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —Г—А–Њ–≤–љ—П —Н–љ–µ—А–≥–µ—В–Є—З–µ—Б–Ї–Є—Е —В—А–∞—В –≤ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Є–Є —Б —Г—А–Њ–≤–љ–µ–Љ —Н–љ–µ—А–≥–µ—В–Є—З–µ—Б–Ї–Є—Е —Б—Г–±—Б—В—А–∞—В–Њ–≤, –Є–Љ–µ—О—Й–Є—Е—Б—П –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ (Landsberg L., 1986). –Ю–і–љ–∞–Ї–Њ –≤ —Г—Б–ї–Њ–≤–Є—П—Е –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є –≥–Є–њ–µ—А–∞–Ї—В–Є–≤–Є–Ј–∞—Ж–Є—П –°–Э–° –њ—А–Є–≤–Њ–і–Є—В –Ї –њ–Њ—П–≤–ї–µ–љ–Є—О –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є –Ј–∞ —Б—З–µ—В —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є —Б—В–Є–Љ—Г–ї—П—Ж–Є—П —Б–µ—А–і—Ж–∞, —Б–Њ—Б—Г–і–Њ–≤ –Є –њ–Њ—З–µ–Ї (—А–Є—Б. 2).
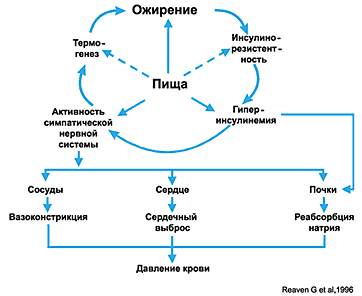
–†–Є—Б. 2. –Т–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –Љ–µ–ґ–і—Г –Є–љ—Б—Г–ї–Є–љ–Њ–Љ –Є —Г—А–Њ–≤–љ–µ–Љ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П —Г –±–Њ–ї—М–љ—Л—Е —Б –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–µ–є, —Б–≤—П–Ј–∞–љ–љ–Њ–є —Б –Њ–ґ–Є—А–µ–љ–Є–µ–Љ
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –∞—А—В–µ—А–Є–∞–ї—М–љ–∞—П –≥–Є–њ–µ—А—В–Њ–љ–Є—П –њ—А–Є –Њ–ґ–Є—А–µ–љ–Є–Є —П–≤–ї—П–µ—В—Б—П —Б–≤–Њ–µ–≥–Њ —А–Њ–і–∞ –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л–Љ –њ–Њ–±–Њ—З–љ—Л–Љ –њ—А–Њ–і—Г–Ї—В–Њ–Љ –і–µ–є—Б—В–≤–Є—П –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л—Е –љ–∞ –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є–µ —Н–љ–µ—А–≥–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –±–∞–ї–∞–љ—Б–∞ –Є —Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є –≤–µ—Б–∞ —В–µ–ї–∞.–†–µ–Ј—Г–ї—М—В–∞—В—Л —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ–і—В–≤–µ—А–і–Є–ї–Є: –Њ—Б—В—А–Њ–µ –Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Є–љ—Б—Г–ї–Є–љ–∞ –≤ –Ї—А–Њ–≤–Є —Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –°–Э–° –Є –њ–Њ–≤—Л—И–∞–µ—В –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—О –Ї–∞—В–µ—Е–Њ–ї–∞–Љ–Є–љ–Њ–≤ –≤ –Ї—А–Њ–≤–Є (Young J.B., 1988; Landsberg L., 1990; Bunag R.D. et al., 1991). –Ф–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л–µ –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М—Б—В–≤–∞, –њ–Њ–і—В–≤–µ—А–ґ–і–∞—О—Й–Є–µ —Б—В–Є–Љ—Г–ї–Є—А—Г—О—Й–Є–є —Н—Д—Д–µ–Ї—В –Є–љ—Б—Г–ї–Є–љ–∞ –љ–∞ —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М, –њ–Њ–ї—Г—З–µ–љ—Л –њ—А–Є –Є—Б–њ—Л—В–∞–љ–Є—П—Е —Б–Њ–Љ–∞—В–Њ—Б—В–∞—В–Є–љ–∞. –≠—В–Њ—В –≥–Њ—А–Љ–Њ–љ, —Г–≥–љ–µ—В–∞—О—Й–Є–є —Н–љ–і–Њ–≥–µ–љ–љ—Г—О —Б–µ–Ї—А–µ—Ж–Є—О –Є–љ—Б—Г–ї–Є–љ–∞, –њ—А–Є–≤–Њ–і–Є—В –Ї —Б–љ–Є–ґ–µ–љ–Є—О –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–є –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –≤ –њ–ї–∞–Ј–Љ–µ –Є —Г–Љ–µ–љ—М—И–µ–љ–Є—О —Г—А–Њ–≤–љ—П –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П —Г –±–Њ–ї—М–љ—Л—Е —Б –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–µ–є (Supiano M.A. et al., 1992; Carretta R. et al., 1989). –Я—А–Є –љ–µ—Д–∞—А–Љ–∞–Ї–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П—Е, —Г–Љ–µ–љ—М—И–∞—О—Й–Є—Е –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є (–Њ–≥—А–∞–љ–Є—З–µ–љ–Є–µ –Ї–∞–ї–Њ—А–Є–є, —Б–љ–Є–ґ–µ–љ–Є–µ –≤–µ—Б–∞, —Д–Є–Ј–Є—З–µ—Б–Ї–Є–µ —Г–њ—А–∞–ґ–љ–µ–љ–Є—П), —В–∞–Ї–ґ–µ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ –њ–∞—А–∞–ї–ї–µ–ї—М–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Є–љ—Б—Г–ї–Є–љ–∞ –≤ –Ї—А–Њ–≤–Є, –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Є —Г—А–Њ–≤–љ—П –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П (Jung R.T. et al., 1979; Stumler R. et al., 1987; Krotkiewski M. et al., 1979; Tuck M.L., 1992).
–Ф–∞–љ–љ—Л–µ —Н–њ–Є–і–µ–Љ–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ–і—В–≤–µ—А–і–Є–ї–Є –Ј–љ–∞—З–Є–Љ–Њ—Б—В—М –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–Є –Є –∞–Ї—В–Є–≤–∞—Ж–Є–Є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –≤ —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–Є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є –љ–∞ –њ–Њ–њ—Г–ї—П—Ж–Є–Њ–љ–љ–Њ–Љ —Г—А–Њ–≤–љ–µ. –Т Normative Aging Study, –њ—А–Њ–≤–µ–і–µ–љ–љ–Њ–Љ –≤ –С–Њ—Б—В–Њ–љ–µ, –°–®–Р, —Г —Г—З–∞—Б—В–љ–Є–Ї–Њ–≤ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Њ—Ж–µ–љ–Є–≤–∞–ї–∞—Б—М –њ–Њ —Г—А–Њ–≤–љ—О —Б—Г—В–Њ—З–љ–Њ–є —Н–Ї—Б–Ї—А–µ—Ж–Є–Є –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ —Б –Љ–Њ—З–Њ–є. –С—Л–ї–Њ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ, —З—В–Њ —Н—В–∞ –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –њ—А—П–Љ–Њ –Ї–Њ—А—А–µ–ї–Є—А—Г–µ—В —Б –Є–љ–і–µ–Ї—Б–Њ–Љ –Љ–∞—Б—Б—Л —В–µ–ї–∞ –Є –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ –њ–Њ–≤—Л—И–µ–љ–∞ —Г –ї–Є—Ж —Б –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–µ–є (Troisi R.J. et al., 1991). –°—В–Є–Љ—Г–ї–Є—А—Г—О—Й–Є–є —Н—Д—Д–µ–Ї—В –Є–љ—Б—Г–ї–Є–љ–∞ –≤ –Њ—В–љ–Њ—И–µ–љ–Є–Є –°–Э–° –њ—А–Є –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–Є –љ–µ –Њ—В–ї–Є—З–∞–µ—В—Б—П –Њ—В —В–∞–Ї–Њ–≤–Њ–≥–Њ —Г –ї–Є—Ж —Б –љ–Њ—А–Љ–∞–ї—М–љ–Њ–є —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В—М—О –Ї –Є–љ—Б—Г–ї–Є–љ—Г. –Я–Њ–Ј–Є—В–Є–≤–љ–∞—П –∞—Б—Б–Њ—Ж–Є–∞—Ж–Є—П –Љ–µ–ґ–і—Г —Г—А–Њ–≤–љ–µ–Љ –Є–љ—Б—Г–ї–Є–љ–∞ –њ–ї–∞–Ј–Љ—Л, —Б—Г—В–Њ—З–љ–Њ–є —Н–Ї—Б–Ї—А–µ—Ж–Є–Є –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –Є —Г—А–Њ–≤–љ–µ–Љ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П, –Њ–±–љ–∞—А—Г–ґ–µ–љ–љ–∞—П –≤ —Н—В–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є, —Б–Њ—Е—А–∞–љ—П–ї–∞—Б—М –њ—А–Є —Г—З–µ—В–µ –≤–ї–Є—П–љ–Є—П –љ–∞ —Н—В–Є –њ–∞—А–∞–Љ–µ—В—А—Л –Є–љ–і–µ–Ї—Б–∞ –Љ–∞—Б—Б—Л —В–µ–ї–∞ –Є —В–Є–њ–∞ —А–∞—Б–њ—А–µ–і–µ–ї–µ–љ–Є—П –ґ–Є—А–∞ –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ (O’Hare J.A. et al., 1989).
–†–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ —В–Њ–Љ, —З—В–Њ –Є–љ—Б—Г–ї–Є–љ –Є –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В—М —П–≤–ї—П—О—В—Б—П —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–∞–Љ–Є —Б–Є—Б—В–µ–Љ—Л —А–µ–≥—Г–ї—П—Ж–Є–Є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П –Є –≤–µ–і—Г—Й—Г—О —А–Њ–ї—М –≤ —А–µ–∞–ї–Є–Ј–∞—Ж–Є–Є –Є—Е –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –љ–∞ —Г—А–Њ–≤–µ–љ—М –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П –Є–≥—А–∞–µ—В —Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–∞—П –∞–Ї—В–Є–≤–Є–Ј–∞—Ж–Є—П —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л (Landsberg L., 1985).
–Т 1992–95 –≥–Њ–і–∞—Е Young –Є MacDonald –Њ–±–Њ–±—Й–Є–ї–Є –Є–Љ–µ—О—Й–Є–µ—Б—П –і–∞–љ–љ—Л–µ –њ–Њ –≤–Њ–њ—А–Њ—Б—Г –Њ —Б–Њ—Б—В–Њ—П–љ–Є–Є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –њ—А–Є –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–Є –Є –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є, —Б–≤—П–Ј–∞–љ–љ–Њ–є —Б –Њ–ґ–Є—А–µ–љ–Є–µ–Љ (Young J.B., MacDonald I.A.,1992; MacDonald I.A., 1995). –Ю–Ї–∞–Ј–∞–ї–Њ—Б—М, —З—В–Њ —Г –ї–Є—Ж —Б –Њ–ґ–Є—А–µ–љ–Є–µ–Љ, –њ–Њ –і–∞–љ–љ—Л–Љ 14 –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –≤ –Ї—А–Њ–≤–Є –±—Л–ї–∞ –љ–Є–ґ–µ, —З–µ–Љ —Г —Е—Г–і—Л—Е; –њ–Њ –і–∞–љ–љ—Л–Љ 21 –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П – –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ —Г –ї–Є—Ж —Б –Є–Ј–±—Л—В–Њ—З–љ—Л–Љ –≤–µ—Б–Њ–Љ –Є –±–µ–Ј –Њ–ґ–Є—А–µ–љ–Є—П –±—Л–ї–Є –Њ–і–Є–љ–∞–Ї–Њ–≤—Л–Љ–Є –Є –њ–Њ –і–∞–љ–љ—Л–Љ 11 –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –њ—А–Є –Њ–ґ–Є—А–µ–љ–Є–Є –±—Л–ї–∞ –≤—Л—И–µ, —З–µ–Љ —Г —Е—Г–і—Л—Е –ї–Є—Ж.
–Я—А–Њ—В–Є–≤–Њ—А–µ—З–Є–≤–Њ—Б—В—М –Є–Љ–µ—О—Й–Є—Е—Б—П —Б–≤–µ–і–µ–љ–Є–є –Њ–± –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –°–Э–° –њ—А–Є –Њ–ґ–Є—А–µ–љ–Є–Є –≤–Њ –Љ–љ–Њ–≥–Њ–Љ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–∞ –Љ–µ—В–Њ–і–∞–Љ–Є, –њ—А–Є–Љ–µ–љ—П–µ–Љ—Л–Љ–Є –і–ї—П –Њ—Ж–µ–љ–Ї–Є —Б–Њ—Б—В–Њ—П–љ–Є—П —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л (Kaasb S. et al., 1995; Astrup A., MacDonald I.A., 1998).
–Т –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ –≤—Л—И–µ –њ—А–Є–≤–µ–і–µ–љ–љ—Л—Е —А–∞–±–Њ—В (Young J.B., MacDonald I.A.,1992; MacDonald I.A., 1995) –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –°–Э–° –Њ—Ж–µ–љ–Є–≤–∞–ї–∞—Б—М –њ–Њ —Г—А–Њ–≤–љ—О –Ї–∞—В–µ—Е–Њ–ї–∞–Љ–Є–љ–Њ–≤ –≤ –Ї—А–Њ–≤–Є –Є–ї–Є –Љ–Њ—З–µ. –Т–Њ—Б–њ—А–Њ–Є–Ј–≤–Њ–і–Є–Љ–Њ—Б—В—М —Н—В–Є—Е –∞–љ–∞–ї–Є–Ј–Њ–≤ –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ –≤—Л—Б–Њ–Ї–∞. –Ъ —В–Њ–Љ—Г –ґ–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –≤ –њ–ї–∞–Ј–Љ–µ –Є –Љ–Њ—З–µ –Ј–∞–≤–Є—Б—П—В –љ–µ —В–Њ–ї—М–Ї–Њ –Њ—В –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Є—Е –љ–µ–є—А–Њ–љ–Њ–≤, –љ–Њ –Є –Њ—В –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є—П –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –Є–Ј –њ—А–Њ—Б–≤–µ—В–Њ–≤ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е —Б–Є–љ–∞–њ—Б–Њ–≤ –≤ –Ї—А–Њ–≤—М –Є –Њ—В —Б–Ї–Њ—А–Њ—Б—В–Є –Є—Б—З–µ–Ј–љ–Њ–≤–µ–љ–Є—П –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –Є–Ј —Ж–Є—А–Ї—Г–ї–Є—А—Г—О—Й–µ–є –Ї—А–Њ–≤–Є, –Ї–∞–ґ–і—Л–є –Є–Ј –Ї–Њ—В–Њ—А—Л—Е –Љ–Њ–ґ–µ—В —И–Є—А–Њ–Ї–Њ –≤–∞—А—М–Є—А–Њ–≤–∞—В—М.
–Т–љ–µ–і—А–µ–љ–Є–µ —В–µ—Е–љ–Є–Ї–Є «–Љ–µ—З–µ–љ—Л—Е» –∞—В–Њ–Љ–Њ–≤ –Є –Љ–Є–Ї—А–Њ–љ–µ–є—А–Њ–≥—А–∞—Д–Є–Є –і–ї—П –њ—А—П–Љ–Њ–≥–Њ –Є–Ј–Љ–µ—А–µ–љ–Є—П –Њ–±–Љ–µ–љ–∞ –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –≤ –Њ–Ї–Њ–љ—З–∞–љ–Є—П—Е –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Њ –≤ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–є –Љ–µ—А–µ —А–∞—Б—И–Є—А–Є—В—М —Б—Г—Й–µ—Б—В–≤—Г—О—Й–Є–µ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Є—П –Њ —Б–Њ—Б—В–Њ—П–љ–Є–Є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л —Г –ї–Є—Ж —Б –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–µ–є –Є –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В—М—О (Astrup A., MacDonald I.A., 1998; Grassi G., Esler M., 1999).
–Р–Ї—В–Є–≤–∞—Ж–Є–Є —Ж–µ–љ—В—А–∞–ї—М–љ—Л—Е –Њ—В–і–µ–ї–Њ–≤ –°–Э–° –Є–Ј–Љ–µ–љ—П–µ—В –Њ–±–Љ–µ–љ –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –≤ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –Њ–Ї–Њ–љ—З–∞–љ–Є—П—Е –°–Э–°. –Т–Њ –Љ–љ–Њ–≥–Є—Е —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Б–Є—В—Г–∞—Ж–Є—П—Е –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–∞—П —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–∞—П –љ–µ—А–≤–љ–∞—П —Б–Є—Б—В–µ–Љ–∞ —А–µ–∞–≥–Є—А—Г–µ—В –љ–µ –Ї–∞–Ї –µ–і–Є–љ–Њ–µ —Ж–µ–ї–Њ–µ, –∞ –Ї–∞–Ї —Б–Њ—З–µ—В–∞–љ–Є–µ –Њ—В–і–µ–ї—М–љ—Л—Е —З–∞—Б—В–µ–є, –Є–Љ–µ—О—Й–Є—Е –≤—Л—А–∞–ґ–µ–љ–љ—Л–µ —А–µ–≥–Є–Њ–љ–∞–ї—М–љ—Л–µ –Њ—В–ї–Є—З–Є—П –њ–Њ —Б—В–µ–њ–µ–љ–Є —А–∞–Ј–≤–Є–≤–∞—О—Й–µ–є—Б—П —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–∞—Ж–Є–Є (van Baak M.A., 2001).
–Ш–Ј–Љ–µ—А–µ–љ–Є—П —А–µ–≥–Є–Њ–љ–∞–ї—М–љ–Њ–≥–Њ –≤—Л–і–µ–ї–µ–љ–Є—П –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Є —Б–і–µ–ї–∞—В—М –≤—Л–≤–Њ–і –Њ —В–Њ–Љ, —З—В–Њ –≤ —Г—Б–ї–Њ–≤–Є—П—Е –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–Є –Є –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є –њ—А–Є –Њ–ґ–Є—А–µ–љ–Є–Є –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –њ–Њ–≤—Л—И–µ–љ–∞ –≤ –њ–Њ—З–Ї–∞—Е –Є —Б–љ–Є–ґ–µ–љ–∞ –≤ —Б–µ—А–і—Ж–µ (Vaz M. et al., 1997; Rumantir M.S. et al., 1999; Kaasb S. et al., 1995). –Я—А–Є —Н—В–Њ–Љ —Г—А–Њ–≤–µ–љ—М —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –≤ —Б–Ї–µ–ї–µ—В–љ–Њ–є –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А–µ —В–µ–Љ –≤—Л—И–µ, —З–µ–Љ –±–Њ–ї—М—И–µ –≤–µ—Б —В–µ–ї–∞, –Є–љ–і–µ–Ї—Б –Љ–∞—Б—Б—Л —В–µ–ї–∞ –Є –њ—А–Њ—Ж–µ–љ—В –ґ–Є—А–∞ –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ (Spraul M. et al., 1993; Scherrer U. et al., 1994; Vollenweider P. et al., 1994; Tataranni P.A. et al., 1999). –Я–Њ—З–µ—З–љ–∞—П –≥–Є–њ–µ—А—Б–Є–Љ–њ–∞—В–Є–Ї–Њ—В–Њ–љ–Є—П —Г—Б–Є–ї–Є–≤–∞–µ—В –Ј–∞–і–µ—А–ґ–Ї—Г –љ–∞—В—А–Є—П, –ґ–Є–і–Ї–Њ—Б—В–Є, –Є–Ј–Љ–µ–љ—П–µ—В –њ–Њ—З–µ—З–љ—Г—О –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є–Ї—Г, —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В –≤—Л–і–µ–ї–µ–љ–Є–µ —А–µ–љ–Є–љ–∞, –њ—А–Є–≤–Њ–і—П –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ –Ї –њ–Њ–≤—Л—И–µ–љ–Є—О –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П.
–Ю—Б–Њ–±–∞—П —А–Њ–ї—М –њ–Њ—З–µ–Ї –≤ —А–∞–Ј–≤–Є—В–Є–Є –Є–љ—Б—Г–ї–Є–љ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є –±—Л–ї–∞ –Њ—Б–Њ–Ј–љ–∞–љ–∞ —Б–Њ–≤—Б–µ–Љ –љ–µ–і–∞–≤–љ–Њ. –†–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ –і–≤—Г—Б—В–Њ—А–Њ–љ–љ—П—П –і–µ–Є–љ–µ—А–≤–∞—Ж–Є—П –њ–Њ—З–µ–Ї —Г –Ї—А—Л—Б –њ—А–µ–і–Њ—В–≤—А–∞—Й–∞–µ—В –±—Л—Б—В—А—Л–є –њ–Њ–і—К–µ–Љ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П –≤–Њ –≤—А–µ–Љ—П –Є–љ—Д—Г–Ј–Є–Є –Є–љ—Б—Г–ї–Є–љ–∞ –Є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ —Б–љ–Є–ґ–∞–µ—В —Б—Д–Њ—А–Љ–Є—А–Њ–≤–∞–≤—И—Г—О—Б—П –≥–Є–њ–µ—А—В–Њ–љ–Є—О, –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ—Г—О –і–ї–Є—В–µ–ї—М–љ—Л–Љ –≤–≤–µ–і–µ–љ–Є–µ–Љ –Є–љ—Б—Г–ї–Є–љ–∞ (Wann–Chu Huang et al., 1998). –Ф–µ–Є–љ–µ—А–≤–∞—Ж–Є—П –њ–Њ—З–µ–Ї —Б–љ–Є–ґ–∞–µ—В –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –≥–Є–њ–µ—А—В–Њ–љ–Є–Є –Є —Г —Б–Њ–±–∞–Ї —Б –Њ–ґ–Є—А–µ–љ–Є–µ–Љ –Є –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–µ–є (Kaasb S. et al., 1995). –≠—В–Є —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –і–∞–љ–љ—Л–µ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ —В–Њ–Љ, —З—В–Њ –љ–∞–ї–Є—З–Є–µ –Є–љ—В–∞–Ї—В–љ–Њ–є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –њ–Њ—З–µ—З–љ–Њ–є –Є–љ–љ–µ—А–≤–∞—Ж–Є–Є —П–≤–ї—П–µ—В—Б—П –Њ–±—П–Ј–∞—В–µ–ї—М–љ—Л–Љ —Г—Б–ї–Њ–≤–Є–µ–Љ —А–∞–Ј–≤–Є—В–Є—П –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є, —Б–≤—П–Ј–∞–љ–љ–Њ–є —Б —Г—Б—В–Њ–є—З–Є–≤–Њ–є –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–µ–є.
–Э–µ—А–≤—Л –њ–Њ—З–µ–Ї –Є–≥—А–∞—О—В –≤–∞–ґ–љ–µ–є—И—Г—О —А–Њ–ї—М –≤ –Ї–Њ–љ—В—А–Њ–ї–µ –њ–Њ—З–µ—З–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є (DiBona G.F., 1989). –•–Њ—А–Њ—И–Њ –Є–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –њ–Њ—З–µ—З–љ–∞—П —А–µ–≥—Г–ї—П—Ж–Є—П –±–∞–ї–∞–љ—Б–∞ –ґ–Є–і–Ї–Њ—Б—В–Є –Є –љ–∞—В—А–Є—П –Є–≥—А–∞–µ—В —А–µ—И–∞—О—Й—Г—О —А–Њ–ї—М –≤ –і–Њ–ї–≥–Њ—Б—А–Њ—З–љ–Њ–Љ –Ї–Њ–љ—В—А–Њ–ї–µ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П –≤ –љ–Њ—А–Љ–µ –Є –њ—А–Є –њ–∞—В–Њ–ї–Њ–≥–Є–Є (Guyton A.C. et al., 1991). C—В–Є–Љ—Г–ї—П—Ж–Є—П —Н—Д—Д–µ—А–µ–љ—В–љ—Л—Е (—Ж–µ–љ—В—А–Њ–±–µ–ґ–љ—Л—Е) –љ–µ—А–≤–Њ–≤ –њ–Њ—З–µ–Ї –Є–Ј–Љ–µ–љ—П–µ—В –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є–Ї—Г –њ–Њ—З–µ–Ї, —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В –Ї–∞–љ–∞–ї—М—Ж–µ–≤—Г—О —А–µ–∞–±—Б–Њ—А–±—Ж–Є—О –љ–∞—В—А–Є—П (DiBona G.F.,1989; Moss N.G., 1982).
–°–Њ–≤—Б–µ–Љ –љ–µ–і–∞–≤–љ–Њ –±—Л–ї–Њ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ, —З—В–Њ –њ—А–Є —Б–∞—Е–∞—А–љ–Њ–Љ –і–Є–∞–±–µ—В–µ 2 —В–Є–њ–∞ —Б—Г—Й–µ—Б—В–≤—Г–µ—В –µ—Й–µ –Њ–і–Є–љ –Љ–µ—Е–∞–љ–Є–Ј–Љ, –њ—А–Є–≤–Њ–і—П—Й–Є–є –Ї —Г—Б–Є–ї–µ–љ–Є—О –њ–Њ—З–µ—З–љ–Њ–є –≥–Є–њ–µ—А—Б–Є–Љ–њ–∞—В–Є–Ї–Њ—В–Њ–љ–Є–Є. –Ю–љ —Б–≤—П–Ј–∞–љ —Б –≤–ї–Є—П–љ–Є–µ–Љ –≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–Є –љ–∞ —Н–Ї—Б–њ—А–µ—Б—Б–Є—О –≥–µ–љ–∞ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≥–µ–љ–∞ –≤ –њ–Њ—З–µ—З–љ–Њ–є —В–Ї–∞–љ–Є –≤ —Г—Б–ї–Њ–≤–Є—П—Е –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є.
–Т –љ–Њ—А–Љ–µ –Є–љ—Б—Г–ї–Є–љ –њ–Њ–і–∞–≤–ї—П–µ—В —Б—В–Є–Љ—Г–ї–Є—А—Г—О—Й–Є–є —Н—Д—Д–µ–Ї—В –≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–Є –љ–∞ —Н–Ї—Б–њ—А–µ—Б—Б–Є—О –≥–µ–љ–∞ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≥–µ–љ–∞ –≤ –Ї–ї–µ—В–Ї–∞—Е –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤ –њ–Њ—З–µ–Ї –Є –њ—А–µ–њ—П—В—Б—В–≤—Г–µ—В —Г–≤–µ–ї–Є—З–µ–љ–Є—О —Б–µ–Ї—А–µ—Ж–Є–Є –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≥–µ–љ–∞ (—А–Є—Б. 3). –Я—А–Є —Б–∞—Е–∞—А–љ–Њ–Љ –і–Є–∞–±–µ—В–µ 2 —В–Є–њ–∞ –≤ —Б–≤—П–Ј–Є —Б –љ–∞–ї–Є—З–Є–µ–Љ –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є –њ–Њ–і–∞–≤–ї–µ–љ–Є–µ –Є–љ—Б—Г–ї–Є–љ–Њ–Љ –≥–ї—О–Ї–Њ–Ј–Њ–—Б—В–Є–Љ—Г–ї–Є—А—Г–µ–Љ–Њ–є —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є –≥–µ–љ–∞ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≥–µ–љ–∞ –≤ –Ї–ї–µ—В–Ї–∞—Е –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤ –њ–Њ—З–µ–Ї –љ–µ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В, —Н–Ї—Б–њ—А–µ—Б—Б–Є—П –≥–µ–љ–∞ —А–∞—Б—В–Њ—А–Љ–∞–ґ–Є–≤–∞–µ—В—Б—П –Є —Б–µ–Ї—А–µ—Ж–Є—П –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≥–µ–љ–∞ —Г—Б–Є–ї–Є–≤–∞–µ—В—Б—П (Zang S.L. et al., 2002). –Я–Њ––≤–Є–і–Є–Љ–Њ–Љ—Г, –Є–Љ–µ–љ–љ–Њ —Н—В–Њ—В –Љ–µ—Е–∞–љ–Є–Ј–Љ –ї–µ–ґ–Є—В –≤ –Њ—Б–љ–Њ–≤–µ –Њ–±–љ–∞—А—Г–ґ–µ–љ–љ–Њ–≥–Њ —Г–≤–µ–ї–Є—З–µ–љ–Є—П –њ—А–Њ–і—Г–Ї—Ж–Є–Є –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–∞ II –≤ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤—Л—Е –Є –Ї–∞–љ–∞–ї—М—Ж–µ–≤—Л—Е –Ї–ї–µ—В–Ї–∞—Е –њ–Њ—З–µ—З–љ–Њ–є —В–Ї–∞–љ–Є –њ–Њ–і –≤–ї–Є—П–љ–Є–µ–Љ –≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–Є (Burns K.D., 2000).

–†–Є—Б. 3. –Я–Њ–і–∞–≤–ї–µ–љ–Є–µ –Є–љ—Б—Г–ї–Є–љ–Њ–Љ —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є –≥–µ–љ–∞ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≥–µ–љ–∞
–Т–љ—Г—В—А–Є–њ–Њ—З–µ—З–љ—Л–є –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ II —Б–≤—П–Ј—Л–≤–∞–µ—В—Б—П —Б –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–Љ–Є –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ I —А–µ—Ж–µ–њ—В–Њ—А–∞–Љ–Є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤ –Є –Ї–∞–љ–∞–ї—М—Ж–µ–≤ –њ–Њ—З–µ–Ї, —Б —А–µ—Ж–µ–њ—В–Њ—А–∞–Љ–Є –µ–µ —Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–µ—В–Є –Є –Ї–ї–µ—В–Њ–Ї –Є–љ—В–µ—А—Б—В–Є—Ж–Є—П (Burns KD, 2000). –Ч–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –Р–Ґ1 —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –Њ—В–Љ–µ—З–µ–љ–Њ –њ—А–Є —Б–∞—Е–∞—А–љ–Њ–Љ –і–Є–∞–±–µ—В–µ 2 —В–Є–њ–∞.–Т–Њ–Ј–і–µ–є—Б—В–≤–Є–µ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–∞ II –љ–∞ —А–µ—Ж–µ–њ—В–Њ—А—Л –Р–Ґ1, –Ї–Њ–љ—В—А–Њ–ї–Є—А—Г—О—Й–Є–µ –≤—Л–і–µ–ї–µ–љ–Є–µ –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –≤ —Б–Є–љ–∞–њ—Б–∞—Е —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –њ–Њ—З–µ–Ї, –≤–µ–і–µ—В –Ї –љ–∞—А–∞—Б—В–∞–љ–Є—О —А–µ–љ–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—Б–Є–Љ–њ–∞—В–Є–Ї–Њ—В–Њ–љ–Є–Є (—А–Є—Б. 4). –Ь–µ–і–Є–Ї–∞–Љ–µ–љ—В–Њ–Ј–љ–Њ–µ –±–ї–Њ–Ї–Є—А–Њ–≤–∞–љ–Є–µ —Н—В–Њ–≥–Њ –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞ –њ—А–µ–њ—П—В—Б—В–≤—Г–µ—В —А–∞–Ј–≤–Є—В–Є—О –Є–љ—Б—Г–ї–Є–љ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є (Te–Chao Fang, Wann–Chu Huang, 1998).

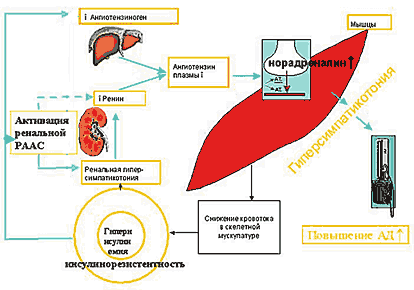
–†–Є—Б. 4. –°—Е–µ–Љ–∞ –∞–Ї—В–Є–≤–∞—Ж–Є–Є –њ–Њ—З–µ—З–љ–Њ–є –†–Р–Р–° –њ—А–Є —Б–∞—Е–∞—А–љ–Њ–Љ –і–Є–∞–±–µ—В–µ 2 —В–Є–њ–∞
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ —А–µ–љ–∞–ї—М–љ–∞—П –≥–Є–њ–µ—А—Б–Є–Љ–њ–∞—В–Є–Ї–Њ—В–Њ–љ–Є—П, —П–≤–ї—П—П—Б—М —Е–∞—А–∞–Ї—В–µ—А–љ–Њ–є –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—М—О –Є–љ—Б—Г–ї–Є–љ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є, –≤–Њ–Ј–љ–Є–Ї–∞–µ—В, –≤–Њ––њ–µ—А–≤—Л—Е, –Ї–∞–Ї –њ–Њ—Б–ї–µ–і—Б—В–≤–Є–µ –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є—З–µ—Б–Ї–Њ–є —Б—В–Є–Љ—Г–ї—П—Ж–Є–Є —Ж–µ–љ—В—А–∞–ї—М–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Є, –≤–Њ––≤—В–Њ—А—Л—Е, –Ї–∞–Ї —А–µ–Ј—Г–ї—М—В–∞—В —Г–≤–µ–ї–Є—З–µ–љ–Є—П –≤—Л–і–µ–ї–µ–љ–Є—П –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –≤ —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Є—Е —Б–Є–љ–∞–њ—Б–∞—Е –њ–Њ—З–µ–Ї –≤—Б–ї–µ–і—Б—В–≤–Є–µ –∞–Ї—В–Є–≤–Є–Ј–∞—Ж–Є—П –њ–Њ—З–µ—З–љ–Њ–є —В–Ї–∞–љ–µ–≤–Њ–є —А–µ–љ–Є–љ––∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≤–Њ–є —Б–Є—Б—В–µ–Љ—Л –≤ —Г—Б–ї–Њ–≤–Є—П—Е –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є.–У–Є–њ–µ—А—Б–Є–Љ–њ–∞—В–Є–Ї–Њ—В–Њ–љ–Є—П —Г—Б–Є–ї–Є–≤–∞–µ—В —Б–µ–Ї—А–µ—Ж–Є—О —А–µ–љ–Є–љ–∞ –≤ –њ–Њ—З–Ї–∞—Е (Di B–Њna G.F.,1989; Moss M.G., 1982). –Я–Њ–≤—Л—И–µ–љ–Є–µ —А–µ–љ–Є–љ–∞ –∞–Ї—В–Є–≤–Є–Ј–Є—А—Г–µ—В —А–µ–љ–Є–љ––∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ––∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ–Њ–≤—Г—О —Б–Є—Б—В–µ–Љ—Г (–†–Р–Р–°). –Ь–љ–Њ–≥–Є–µ –∞–≤—В–Њ—А—Л –Њ—В–Љ–µ—З–∞—О—В –њ–Њ–≤—Л—И–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —А–µ–љ–Є–љ––∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≤–Њ–є —Б–Є—Б—В–µ–Љ—Л –њ—А–Є –Ї–Њ–Љ–њ–µ–љ—Б–Є—А–Њ–≤–∞–љ–љ–Њ–Љ —Б–∞—Е–∞—А–љ–Њ–Љ –і–Є–∞–±–µ—В–µ (Mann J., Ritz E., 1988; Miller J.A., 1999; Brands M.W., Fitzgerald S.M., 2001).
–£–≤–µ–ї–Є—З–µ–љ–Є–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–∞ II –≤–Њ–Ј–і–µ–є—Б—В–≤—Г–µ—В –љ–∞ —А–µ—Ж–µ–њ—В–Њ—А—Л —А–µ–Ј–Є—Б—В–Є–≤–љ—Л—Е —Б–Њ—Б—Г–і–Њ–≤ –Є –љ–∞ –Р–Ґ1 —А–µ—Ж–µ–њ—В–Њ—А—Л –≤ –љ–µ–є—А–Њ–Љ—Л—И–µ—З–љ—Л—Е —Б–Є–љ–∞–њ—Б–Њ–≤ —Б–Ї–µ–ї–µ—В–љ–Њ–є –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А—Л. –Т —А–µ–Ј—Г–ї—М—В–∞—В–µ –≤–Њ–Ј–љ–Є–Ї–∞–µ—В –њ–Њ–і—К–µ–Љ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П –Є –і–∞–ї—М–љ–µ–є—И–µ–µ –љ–∞—А–∞—Б—В–∞–љ–Є–µ —Б–Є–Љ–њ–∞—В–Є–Ї–Њ—В–Њ–љ–Є–Є –≤ —Б–Ї–µ–ї–µ—В–љ–Њ–є –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А–µ (—А–Є—Б. 5). –Ф–ї–Є—В–µ–ї—М–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ –Љ—Л—И–µ—З–љ–Њ–є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –≤—Л–Ј—Л–≤–∞–µ—В —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –Ї–∞–њ–Є–ї–ї—П—А–љ–Њ–є —Б–µ—В–Є –Љ—Л—И—Ж, —Б–љ–Є–ґ–∞–µ—В –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –Љ–µ–і–ї–µ–љ–љ–Њ —Б–Њ–Ї—А–∞—Й–∞—О—Й–Є—Е—Б—П –Љ—Л—И–µ—З–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ, –Њ–њ—А–µ–і–µ–ї—П—О—Й–Є—Е —Г—А–Њ–≤–µ–љ—М —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є –Њ—А–≥–∞–љ–Є–Ј–Љ–∞ –Ї –Є–љ—Б—Г–ї–Є–љ—Г. –£—Е—Г–і—И–µ–љ–Є–µ –Ї—А–Њ–≤–Њ—В–Њ–Ї–∞ —Б–Ї–µ–ї–µ—В–љ—Л—Е –Љ—Л—И—Ж, —П–≤–ї—П—О—Й–Є—Е—Б—П –≥–ї–∞–≤–љ—Л–Љ –њ–Њ—В—А–µ–±–Є—В–µ–ї–µ–Љ –≥–ї—О–Ї–Њ–Ј—Л –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ, –њ—А–Є–≤–Њ–і–Є—В –Ї –њ–Њ–љ–Є–ґ–µ–љ–Є—О —В—А–∞–љ—Б–њ–Њ—А—В–∞ –≥–ї—О–Ї–Њ–Ј—Л –≤ –Љ—Л—И—Ж–∞—Е –Є –і–∞–ї—М–љ–µ–є—И–µ–Љ—Г –љ–∞—А–∞—Б—В–∞–љ–Є—О –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є –Є –Ї–Њ–Љ–њ–µ–љ—Б–∞—В–Њ—А–љ–Њ–є –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є–Є. (Lind L., Lithell H., 1993; Zeman R.J., et al., 1988; Rupp H., et al., 1991). –§–Њ—А–Љ–Є—А—Г–µ—В—Б—П «–њ–Њ—А–Њ—З–љ—Л–є –Ї—А—Г–≥», –≤–µ–і—Г—Й–Є–є –Ї –≤–Њ–Ј–Њ–±–љ–Њ–≤–ї—П—О—Й–µ–є—Б—П –∞–Ї—В–Є–≤–∞—Ж–Є–Є –≤—Б–µ—Е –≤—Е–Њ–і—П—Й–Є—Е –≤ –љ–µ–≥–Њ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤.
–†–Є—Б. 5. –У–Є–њ–µ—А—Б–Є–Љ–њ–∞—В–Є–Ї–Њ—В–Њ–љ–Є—П –Є –∞—А—В–µ—А–Є–∞–ї—М–љ–∞ –≥–Є–њ–µ—А—В–Њ–љ–Є—П –њ—А–Є –Є–љ—Б—Г–ї–Є–љ–Ј–∞–≤–Є—Б–Є–Љ–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є
–Т –Ј–∞–Ї–ї—О—З–µ–љ–Є–µ –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –µ—Й–µ —А–∞–Ј –≤–µ—А–љ—Г—В—М—Б—П –Ї –≤—Л–≤–Њ–і–∞–Љ United Kingdom Prospective Diabetes Study.–Т–µ—Б—М–Љ–∞ –≤–∞–ґ–љ—Л–Љ –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В—Б—П –≤—Л–≤–Њ–і –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–µ–є –Њ —В–Њ–Љ, —З—В–Њ –Р–Я–§––Є–љ–≥–Є–±–Є—В–Њ—А—Л –Є b––∞–і—А–µ–љ–Њ–±–ї–Њ–Ї–∞—В–Њ—А—Л –≤ –Њ–і–Є–љ–∞–Ї–Њ–≤–Њ–є —Б—В–µ–њ–µ–љ–Є —Б–њ–Њ—Б–Њ–±–љ—Л —Б–љ–Є–ґ–∞—В—М –Љ–Є–Ї—А–Њ– –Є –Љ–∞–Ї—А–Њ—Б–Њ—Б—Г–і–Є—Б—В—Л–µ –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є—П —Г –±–Њ–ї—М–љ—Л—Е —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ 2 —В–Є–њ–∞ (UKPDS 38, 1998). –Я—А–Є–≤–µ–і–µ–љ–љ—Л–µ –≤—Л—И–µ –і–∞–љ–љ—Л–µ –Њ —А–Њ–ї–Є –љ–∞—З–∞–ї—М–љ–Њ–є –∞–Ї—В–Є–≤–∞—Ж–Є–Є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –Є –њ–Њ—Б–ї–µ–і—Г—О—Й–µ–є –∞–Ї—В–Є–≤–∞—Ж–Є–Є —А–µ–љ–Є–љ––∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≤–Њ–є —Б–Є—Б—В–µ–Љ—Л –≤ —А–∞–Ј–≤–Є—В–Є–Є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є —Г –±–Њ–ї—М–љ—Л—Е —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ 2 —В–Є–њ–∞ –≤–њ–Њ–ї–љ–µ –Њ–±—К—П—Б–љ—П—О—В –њ—А–Є—З–Є–љ—Г –њ–Њ–ї—Г—З–µ–љ–Є—П —В–∞–Ї–Є—Е —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤.
–Т–Њ–Ј–Љ–Њ–ґ–љ–Њ, –µ—Б–ї–Є –±—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–ї–Є –≤ —Б–≤–Њ–µ–є —А–∞–±–Њ—В–µ –≥–Є–њ–Њ—В–µ–љ–Ј–Є–≤–љ—Л–µ –њ—А–µ–њ–∞—А–∞—В—Л, –≤–ї–Є—П—О—Й–Є–µ –љ–∞ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –њ–µ—А–≤–Є—З–љ–Њ–є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–∞—Ж–Є–Є –≤ —Ж–µ–љ—В—А–∞–ї—М–љ–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ–µ –њ—А–Є –Є–љ—Б—Г–ї–Є–љ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є, –≤—Л–≤–Њ–і—Л –Љ–Њ–≥–ї–Є –±—Л –±—Л—В—М –Є–љ—Л–Љ–Є.
–Ю—Б–Њ–±–Њ–µ –≤–љ–Є–Љ–∞–љ–Є–µ –≤ –Ї–∞—З–µ—Б—В–≤–µ –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є –Њ–±–Њ—Б–љ–Њ–≤–∞–љ–љ—Л—Е –≥–Є–њ–Њ—В–µ–љ–Ј–Є–≤–љ—Л—Е —Б—А–µ–і—Б—В–≤ –њ—А–Є –Є–љ—Б—Г–ї–Є–љ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є –њ—А–Є–≤–ї–µ–Ї–∞—О—В –∞–≥–Њ–љ–Є—Б—В—Л –Є–Љ–Є–і–∞–Ј–Њ–ї–Є–љ–Њ–≤—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤, –±–ї–Њ–Ї–Є—А—Г—О—Й–Є—Е —Ж–µ–љ—В—А–∞–ї—М–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –∞–Ї—В–Є–≤–∞—Ж–Є–Є –°–Э–° (—А–Є—Б. 6). –Т–µ–і—М —Н—В–Є –њ—А–µ–њ–∞—А–∞—В—Л, —Б–љ–Є–ґ–∞—П —Ж–µ–љ—В—А–∞–ї—М–љ—Г—О –≥–Є–њ–µ—А—Б–Є–Љ–њ–∞—В–Є–Ї–Њ—В–Њ–љ–Є—О, —Б–њ–Њ—Б–Њ–±–љ—Л —Г–Љ–µ–љ—М—И–Є—В—М, –Ї—А–Њ–Љ–µ —В–Њ–≥–Њ, –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –†–Р–Р–° (–њ–Њ –і–∞–љ–љ—Л–Љ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є —А–µ–љ–Є–љ–∞ –Є –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–∞ II) (—В–∞–±–ї. 1) –Є –њ—А–Є–≤–µ—Б—В–Є –Ї —Б–љ–Є–ґ–µ–љ–Є—О –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є (—А–Є—Б. 7) – —Н—В–Є—Е —Е–∞—А–∞–Ї—В–µ—А–љ—Л—Е –њ–Њ—Б–ї–µ–і—Б—В–≤–Є–є –Є–љ—Б—Г–ї–Є–љ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є. –Я–Њ–і –≤–ї–Є—П–љ–Є–µ–Љ —Н—В–Є—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –Є–Ј–Љ–µ–љ—П–µ—В—Б—П —Г—А–Њ–≤–µ–љ—М –≤–љ—Г—В—А–Є–њ–Њ—З–µ—З–љ–Њ–є –≥–Є–њ–µ—А—Б–Є–Љ–њ–∞—В–Є–Ї–Њ—В–Њ–љ–Є–Є –Є –≥–Є–њ–µ—А–∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–µ–Љ–Є–Є. –Ю–± —Н—В–Њ–Љ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Љ–Є–Ї—А–Њ–∞–ї—М–±—Г–Љ–Є–љ–∞ –≤ –Љ–Њ—З–µ (—А–Є—Б. 8) –њ–Њ–і –Є—Е –≤–ї–Є—П–љ–Є–µ–Љ.


–†–Є—Б. 6. –Ч–љ–∞—З–µ–љ–Є–µ –Є–Ј–±–Є—А–∞—В–µ–ї—М–љ–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П –љ–∞ I1 —А–µ—Ж–µ–њ—В–Њ—А—Л –Є–Љ–Є–і–∞–Ј–Њ–ї–Є–љ–∞
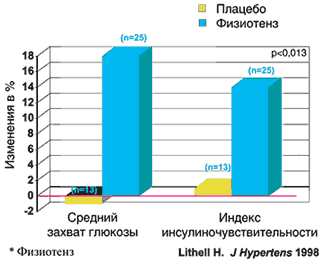
–†–Є—Б. 7. –Т–Њ–Ј–і–µ–є—Б—В–≤–Є–µ —В–µ—А–∞–њ–Є–Є –Љ–Њ–Ї—Б–Њ–љ–Є–і–Є–љ–Њ–Љ* –љ–∞ –≥–ї—О–Ї–Њ–Ј—Г –Є –Є–љ—Б—Г–ї–Є–љ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В—М—О –Є –Р–У
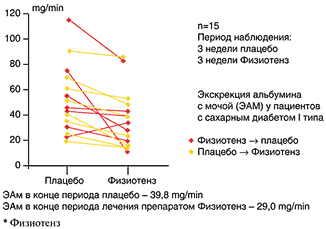
–†–Є—Б. 8. –Э–µ—Д—А–Њ–њ—А–Њ—В–µ–Ї—В–Є–≤–љ—Л–є —Н—Д—Д–µ–Ї—В –Љ–Њ–Ї—Б–Њ–љ–Є–і–Є–љ–∞* —Г –±–Њ–ї—М–љ—Л—Е —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ I —В–Є–њ–∞
–°–љ–Є–ґ–µ–љ–Є–µ —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —П–≤–ї—П–µ—В—Б—П –≤–∞–ґ–љ–µ–є—И–µ–є –Ј–∞–і–∞—З–µ–є –њ—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–Є —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є —Г –±–Њ–ї—М–љ—Л—Е —Б–∞—Е–∞—А–љ—Л–Љ –і–Є–∞–±–µ—В–Њ–Љ 2 —В–Є–њ–∞. –Я—А–µ–њ–∞—А–∞—В—Л, –Ї–Њ—В–Њ—А—Л–µ —Б–њ–Њ—Б–Њ–±–љ—Л –љ–µ —В–Њ–ї—М–Ї–Њ –±–ї–Њ–Ї–Є—А–Њ–≤–∞—В—М —Н—В—Г –∞–Ї—В–Є–≤–љ–Њ—Б—В—М, –љ–Њ –Є –≤–Њ–Ј–і–µ–є—Б—В–≤–Њ–≤–∞—В—М –љ–∞ –Ї–ї—О—З–µ–≤—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л, –µ–µ –Ј–∞–њ—Г—Б–Ї–∞—О—Й–Є–µ –Є –њ–Њ–і–і–µ—А–ґ–Є–≤–∞—О—Й–Є–µ, –љ–µ—Б–Њ–Љ–љ–µ–љ–љ–Њ, –Њ–±–ї–∞–і–∞—О—В –±–Њ–ї—М—И–Є–Љ –њ–Њ—В–µ–љ—Ж–Є–∞–ї–Њ–Љ –њ—А–Є –Є–љ—Б—Г–ї–Є–љ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є –Є –і–Њ—Б—В–Њ–є–љ—Л —В–Њ–≥–Њ –њ—А–Є—Б—В–∞–ї—М–љ–Њ–≥–Њ –≤–љ–Є–Љ–∞–љ–Є—П —Б–Њ —Б—В–Њ—А–Њ–љ—Л –Ї–ї–Є–љ–Є—Ж–Є—Б—В–Њ–≤, –Ї–Њ—В–Њ—А–Њ–µ –Њ—В–Љ–µ—З–∞–µ—В—Б—П —Б–µ–≥–Њ–і–љ—П.
1. Young JB, Landsberg L. Suppression of sympathetic nervous system during fasting. Science 1977; 196: 1473–5.
2. Landsberg L, Young JB.,The influence of diet on the sympathetic nervous system. In: Muller EE, MacLeod RM, Frohman LA, eds. Neuroendocrine perspectives. Vol. 4. Amsterdam: Elservier Science.1985: 191–218.
3. Young JB, Landsberg L. The role of the sympathoadrenal system in modulating energy expenditure. Clin. Endocrinol. Metab. 1984;13:475–99.
4. van Baak MA. The peripheral sympathetic nervous system in humanobesity. Obesityreviews. 2001; 2:3–14.
5. Astrup A, MacDonald IA. Sympathoadrenal system and metabolism. In: Bray GA, Bouchard C, James WPT (eds). Handbook of Obesity Marcel Dekker: New York, 1998, pp491–511.
6. Grassi G, Esler M. How to assess sympathetic activity in humans. J Hypertens 1999; 17: 719–734.
7. Young JB, MacDonald IA. Sympathoadrenal activity in human obesity: heterogeneity of findings since 1980. Int J Obes 1992; 16: 959–967.
8. MacDonald IA. Advances in our undestanding of the role of the sympathetic nervous system in obesity. Int J Obes 1995; 19/7: S2–S7.
9. Spraul M, Ravussin E, Fontvielle AM, Rising R, Larson DE, Anderson EA. Redused sympathetic nervous activity. A potential mechanism predisposing to body weigth gain. J Clin Invest 1993; 92: 1730–1735.
10. Scherrer U, Randin D, Tappy L, Vollenweider P, Jequier E, Nicod P. Body fat and sympathetic nerve activity in healthy subjects. Circulation 1994; 89: 2634–2640.
11. Vollenweider P, Randin D, Tappy L, Jequier E, Nicod D, Scherrer U. Impaired insulin–indused sympathetic neural activity and vasodilatation in skeletal muscle in obese humans. J Clin Invest 1994; 93: 2365–2371.
12. Tataranni PA, Cizza G, Snitker S, Gucciardo F, Lotsikas A, Chrousos GP, Ravussin E. Hypothalamic–pituitary–adrenal axis and sympathetic nervous system activities in Pima Indians and Caucasians. Metabolism 1999; 48: 39
13. Vaz M, Jennings G, Turner A, Cox H, Lambert G, Esler MD. Regional sympathetic nervous activity and oxygen consuption in obese normotensive human subjects. Circulation 1997; 96: 3423–3429.
14. Rumantir MS, Vaz M, Jennings G, Collier G, Kaye DM, Seals DR, Wiesner GH, Brunner–La Rocca HP, Esler MD. Neural mechanisms in human obesity–related hypertansion. J Hypertens 1999; 17: 1125–1133.
15. Reaven GM, Lithell H, Landsberg L. Hypertention and associated metabolic abnormalities – the role of insulin resistance and the sympathoadrenal system. N Engl J Med 1996; 334: 374–381.
16. Skott P, Vaag A, Braun NE et al. Effect of insulin on renal sodium handling in hyperinsulinaemic type 2 (non–insulin–dependent diabetic patiens with peripheral insulin resistance. Diabetologia 1991;34:275–281.
17. Sawicki PT, Heinemann L, Starke A, Berger M. Hyperinsylinaemia is not linked with blood pressure evalution in patient with insulinoma. Diabetologia 1992; 35:649–652.
18. Anderson EA, Mark Al. The vasodilator action of insulin: implication for the insulin hypothesis of hypertention. Hypertention 1993; 21: 136–141. [ Erratum. Hypertention 1993; 21: 745.]
19. Hall JE, Brands MW, Kivlighn SD, Mizelle HL, Hildebrandt A, Gaillard CA. Chronic hyperinsulinemia and blood pressure: interaction with catecholamines? Hypertention 1990; 15:519–527.
20. Brands MW, Hildebrandt DA, Mizelle HL, Hall JE. Sustained Hyperinsulinemia increase arterial pressure in conscious rats. Am J Physiol 1991; 260: R761–R768.
21. Saad MF, Lilloja S, Nyomba BL, et al. Racial differences in the relation between blood pressure and insulin resistance. N Engl J Med 1991; 324:733–739.
22. Falkner B, Hulman S, Kushner H. Insulin–stimulated glucose utilization and bordeline hypertention in young adult blacks.Hypertention 1993; 22:18–25.
23. Tedde R, Sechi LA, Marigliano A, Pala A, Scano L. Antihypertensive effect of insulin reduction in diabetic–hypertensive patients. Am J Hypertens 1989; 2: 163–170.
24. Randeree HA, Omar MAK, Motala AA, Seedat MA. Effect of insulin therapy on blood pressure in NIDDM patients with secondary failure. Diabetes Care 1992; 15:1258–1263.
25. Young JB, Landsberg L. Stimulation of the sympathetic nervous system during sucrose feeding. Nature 1977; 269:615–617.
26. Young JB, Saville E, Rothwell NJ, Stock MJ, Landsberg L. Effect of diet and cold expoure on norepinephrine turnover in brown adipose tissue of the rat. J Clin Invest 1982;69:1061–1071.
27. Schwartz JH, Young JB, Landsberg L. Effect of dietary fat on syparthetic nervous system activity inthe rat. J Clin Invest 1983;72: 361–370.
28. Kaufman LN, Young JB, Landsberg L. Effect of protein on sympathetic nervous system activity in the rat: evidens for nutrient–specific responses. J Clin Invest 1986; 77: 551–558.
29. Landsberg L, Young JB. Insulin–mediated glucose metabolismin the relationship between dietary intake and sympathetic nervous system activity. Int J Obes 1985; 9: Suppl 2: 63–68.
30. Cristlieb AR, Krolewski AS, Warram JH, Soeldner IS. Is insulin the link between hypertention and obesity? Hypertention 1985; 7: Suppl II:II–54–II57.
31. Manicardi V, Camellini L, Bellodi G, Ferrannini E. Evidence for an association of high blood pressure and hyperinsulinrmia in obese man. J Clin Endocrinol Metab 1986; 62:1302–1304.
32. Rose HG, Yalow RS, Schweitzer P, Schwartz E. Insulin as a potential factor influencing blood pressure in amputees. Hypertension 1986; 8: 793–800. [Erratum. Hypertention 1987;9:421.]
33. Modan M, Halkin H, Almog S, et al. Hyperinsulinemia: a link between hypertention obesity and glucose intolerance. J Clin Invest 1985; 75: 809–817.
34. Tappy L, Bovet P, Jequier T, Shamlaye C, Darioli R, Burnand B. Relationship ofof fasting serum insulin concentrations with blood pressure in a representative sample of the adult population of the Seychelles. Int J Obesity 1991; 15: 669–675.
35. Fournier AM, Gadia MT, Kubrusly DB, Skyler JS, Sosenko JM. Blood pressure, insulin, and glycemia in nondiabetic subjects. Am J Med 1986; 80: 861–864.
36. Landsberg L. Diet, obesity, and hypertention: an hypothesis involving insulin, the sympathetic nervous system, and adaptive thermogenesis. Q J Med 1986; 61: 1081–1090.
37. Troisi RJ, Weiss ST, Parker DR, Sparrow D, Young JB, Landsberg L.Relationship of obesity and diet to sympathetic nervous system activity. Hypertention 1991; 17: 669– 677.
38. O’Hara JA, Minaker KL, Meneilly GS, Rowe JW, Palltta JA, Yuong JB. Effect of insulin on plasma norepinephrine and 3,4–dihydroxyphenylalanine in obese men. Metabolism 1989; 38: 322–329.
39. Carretta R, Fabris B, Fichetti F et al. Reduction of blood pressure in obesehyperinsulinaemic hypertensive patients during somatostatin infusion. J Hypertens Suppl 1989; 7: S196–S197.
40. Supiano MA, Hogikyan RV, Marrow LA, et al. Hypertention and insulin resistance: role of sympathetic nervous system activity. Am j Physiol 1992; 363:E935–E942.
41. Jung RT, Sherry PS, Barrand M, Callingham BA, James WPT. Role of cartecholamines in hypotensive responce to dieting. BMJ 1979;1: 12–13.
42. Stumler R, Stumler J, Grimm R , et al. Nutritional therapy for high blood pressure: final report of a four–year randomized controlled trial – the Hypertention Control Program. JAMA 1987; 257: 1484–1491.
43. Krotiewski M, Mandroukas K, Sjostrom L, Wetterquist H, Bjorntorp P. Effects of long–trem physical training on body fat metabolism and blood pressure in obesity. Metabolism 1979; 28: 650–658.
44. Tuck ML. Obesity, the sympathetic nervous system and essential hypertention. Hypertention 1992; 19: Suppl: I–67–I–77.
45. Hall JE, Coleman TG, Mizelle HL. Does chronic hyperinsulinemia cause hypertention. Am J Hypertens 1989;2: 171–173.
46. Anderson EA, Hoffman BP, Balon TW, Sinkey CA, Mark AL. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J Clin Invest 1991; 87: 2246–2252.
47. Zang SL, Chen X, Hsieh TJ, Leclere M, Henley N, Allidina A, Halle JP, Brunette MG, Filep JC, Tang SS,Ingelfinger JR, Chan JS. Hyperflycemia induces insulin resistance on angiotensinogen gene expression in diabetic rat kidney proximal tubular cells. J Endocrinol 2002; 172 (2): 333–344.
48. Burns KD. Angiotensin II and its receptors in the diabetic kidney. Am J Kidney Dis 2000; 36 (3) : 446–467.
49. Brands MW, Fitzgerald SM. Arterial pressure control at the onset of type I diabetes: the role of nitric oxide and the renin–angiotensin system. Am J Hypertens 2001; 14 (6 Pt 2): 126S–131S.
50. Miller JA. Impact of hyperglycemia on the renin angiotensin system in early human type 1 diabetes mellitus. J Am Soc nephrol 1999; 10(8): 1778–1785.
51. Kubo M, Kijohara Y, Kato I, Iwamoto H, Nakayama K, Hirakata H, Fujishima M. Effect of hyperinsulinemia on renal function in a general Japanese population: the Hisayama study. Kidney Int 1999; 55(6): 2450–2456.
52. Mann J, Ritz E. The renin–angiotensin system in diabetic patients. Klin Wochenschr 1988; 66 (18) : 883–893.
53. Kurokawa K, Silverblatt FJ, Klein KL, Wang MS, Lerner RL. Binding of 125I–insulin to the isolated glomeruli of rat kidney. J Clin Invest 1979; 64(5): 1357–1364.
54. Hsieh PS, Huang WC. Chemical sympathectomy attenuates chronic hyperinsulinemia–indused hypertention in conscious rats. Nutr Metab Cardiovasc Dis. 1993;3:173–178.
55. Tomiyama H, Kushiro T, Abeta H, Kurumatani H, Taguchi H, Kuga N, Saito F, Kobayashi F, Otsuka Y, Kanmatsuse K, Kajwara N. Blood pressure response to hyperinsulinemia in salt–sensitive and salt–resistant rats. Hypertension 1992; 20: 596–600.
56. Meehan WP, Buchanam TA, Hsuech W. Chronic insulin administration elevates blood pressure in rats. Hypertention 1994; 23: 1012–1017.
57. Brum M. Insulin stimulates volume absorbtion in the rabbit proximal convoluted tubule. J Clin Invest 1987; 79: 1104–1109.
58. DeFonzo RA, Cooke C, Andres R, Faloona GR, Davis PJ. The effect of insulin in renal handling of sodium, potassium, calcium and phosphate in man. J Clin Invest 1975;55: 845–855.
59. Bunag RD, Knizan–Aghas D, Iton H. Sympethetic activation by chronic insulin treatment in concious rats. J Pharmacol Exp Ther 1991; 259: 131–138.
60. Young JB. Effect of experimental hyperinsulinemia on sympathetic nervous system activity in rats. Life Sci 1988; 49: 193–200.
61. Landsberg L. Insulin resistance, energy balance and sympathetic nervous system activity. Clin Exp Hypertens. 1990; A12: 817–830.
62. Guyton AC, Hall JE, Coleman TG, Manning RD Jr. The dominant role of the kidney in the long–term regulation of arterial pressure in normal and hypertensive state. In: Laragh LH, Brenner BM, eds. Hypertension: Pathophysiology, Diagnosis, and Management. New York, NY: Raven Press; 1991: 1029–1052.
63. DiBona GF. Neural control of renal function: cardiovascular implications. Hypertension 1989; 13: 539–548.
64. Moss NG. Renal function and renal afferent nerve activity. Am J Physiol 1982; 243: F425–F433.
65. Winternitz Sr, Katholi RE, Oparil S. Role of the renal nerves in the development and maintenance of hypertension in the spontaneously hypertensive rat. J Clin Invest 1980; 66: 971–978.
66. Norman RA, Dzielak DJ. Role of the renal nerves in the onset and maintenance of spontaneous hypertension. Am J Physiol 1982; 243: H284–H288.
67. Diz DI, Nasjletti A, Baer PG. Renal denervation at weaning retards development of hypertention in New Zeland genetically hypertensive rats.Hypertension 1982;4: 361–368.
68. Vari RC, Freeman RH, Davis JO, Sweet WD. Role of renal nerves in rats with low–sodium, one–kidney hypertension. Am J Physiol 1986; 250: H189–H194.
69. Vari RC, Zinn S, Verburg KM, Freeman RH. Renal nerves and the pathogenesis of angiotensin–indused hypertention. Hypertension 1987; 9: 345–349.
70. Kassab S, Kato T, Wilkins C, Chen R, Hall JE,Granger JP. Renal denervation attenuates the sodium retention and hypertension associated with obesity. Hypertension 1995; 25(pt 2): 893–897.
71. Wann–Chu Huang, Te–Chao Fang, Juei–Tang Cheng. Renal denervation prevents and reverses hyperinsulinemia–indused hypertention in rats. Hypertention 1998; 32: 249–254.
72. Keen Hl, Brands MW, Alonso–Calicia M, Hall JE. Chronic adrenergic receptor blockade does not prevent hyperinsulinemia–induced hypertention in rats Am J Hypertens 1996; 9: 1192–1199.
73. Calaresu FR, Kin P, Nakamura H, Sato A. Electrophysiological characteristics of renarenal reflex in the cat. J Physiol (Lond) 1978; 283: 141–154.
74. Hall JE, Brands MW, Kivligha SD, Mizelle HL, Hildebrandt DA, Gaillard CA. Chronic hyperinsulinemia and blood pressure: interaction with catecholamines? Hypertension 1990; 15: 519–527.
75. Brands MW, Hildebrandt DA, Mizelle HL, Hall JE. Hypertention during chronic hyperinsulinemia in rats is not salt sensitive. Hypertension 1992; 19 (suppl I): I–83–I–89.
76. Te–Chao Fang, Wann–Chu Huang. Angiotensin receptor blockade blunts hyperinsulinemia–indused hypertension in rats/ Hypertension 1998; 32: 235–242.
77. Te–Chao Fang, Wann–Chu Huang. Role of angiotensin II in hyperinsulinemia–indused hypertention in rats. J hypertension 1998; 16: 1767– 1774.



