




Введение
На протяжении многих лет вопрос о целесообразности существования одинаковых принципов фармакотерапии больных хронической сердечной недостаточностью (ХСН) различной этиологии вызывает активные дебаты среди клиницистов у нас в стране и за рубежом. Причиной служит определенная противоречивость результатов различных клинических исследований. Тем не менее, согласно последним опубликованным практическим рекомендациям по лечению сердечной недостаточности (СН) Американской ассоциации сердца и Американского кардиологического общества, так же как и рабочей группы по сердечной недостаточности Европейского кардиологического общества, существует единый стандартный фармакологический подход в лечении пациентов с систолической левожелудочковой дисфункцией. Согласно этим рекомендациям, практически все больные с СН, независимо от этиологической причины (редким исключением могут являться лишь обструктивные заболевания сердца), должны находиться на лечении ингибиторами ангиотензинпревращающего фермента (АПФ), причем, как правило, в качестве первой линии терапии. Чем же можно объяснить такой успех данной группы лекарственных препаратов, и существуют ли какие-либо особенности их применения при ХСН вследствие некоронарогенных заболеваний сердца?
Эпидемиология и этиология ХСН
Несмотря на значительное количество данных различных клинических исследований при ХСН, эпидемиологические сведения о причинах СН носят чрезвычайно неоднородный характер (табл. 1). Как следует из данных табл. 1, примерно у 60—70% больных в клинических исследованиях в основе развития СН лежала ИБС. Среди других этиологических причин ХСН превалируют АГ и ДКМП. Несмотря на равнозначность критериев включения (имеется в виду этиология СН), в ряде исследований явно преобладают больные с ИБС, тогда как в других отмечается противоположная картина и значительное число пациентов с ДКМП. Данные отличия могут быть связаны как с различием диагностических критериев, преобладанием определенных популяций больных в узкоспециализированных исследовательских центрах, так и с “исследовательским фактором”. Не исключено, что именно этим может быть объяснен факт значительного увеличения числа больных ДКМП в исследованиях с b-блокаторами бисопрололом и карведилолом, так как, по убеждению многих клиницистов, пациенты с этой патологией имеют особенно благоприятный ответ на данный вид лекарственной терапии. В то же время неишемическая этиология СН в большинстве своем далеко не всегда бывает достаточно верифицирована. В повседневной клинической практике довольно часто данный диагноз может быть установлен лишь на основании отсутствия анамнестических, клинических и электрокардиографических признаков заболевания, без проведения ангиографического исследования коронарных артерий. Безусловно, это в значительной мере (по данным некоторых исследований до 30—40%!) маскирует истинные причины развития СН, и в определенной мере именно это может служить причиной неоднородности популяций в различных клинических исследованиях. На сегодняшний день очевидным фактом может быть преобладание ИБС над другими причинами развития СН. В то же время АГ и ДКМП вносят основной вклад в развитие этой тяжелой патологии при некоронарогенных заболеваниях сердца. Существуют ли какие-либо особенности развития СН при этих нозологиях в отличие от ишемической? Почему ингибиторы АПФ целесообразно использовать при ДКМП? Есть ли какие-либо отличия в их применении и эффективности при ишемической и неишемической этиологии СН? Рассмотрению этих и некоторых других вопросов и будет посвящена данная статья. Основные патофизиологические механизмы развития СН неишемической этиологиии
О патофизиологических основах развития СН неишемической этиологии на сегодняшний день нам известно гораздо меньше в отличие от таковой при ИБС, при которой основными составляющими служат перенесенный инфаркт миокарда, наличие “оглушенного”, “спящего” миокарда и развитие процессов патологического ремоделирования сердца. Термин “кардиомиопатия” по сути своей означает заболевание сердечной мышцы. Для ДКМП наиболее характерным является такая перестройка кардиальных структур, в результате которой происходит расширение полостей сердца с выраженной систолической дисфукцией миокарда. Пусковые моменты, причины развития данного патологического ремоделирования сердца могут быть различны, но патофизиологические механизмы во многом схожи. В своей основе кардиомиопатии имеют изменения на молекулярном уровне, на уровне экспрессии генов, ответственных за продукцию определенных белков. Существуют ли какие-либо специфические места воздействия, с точки зрения известного механизма действия ингибиторов АПФ при этой патологии? Известно по крайней мере 3 основных механизма, лежащих в основе кардиомиопатий, когда в результате изменения экспрессии определенных генов происходит перестройка структуры и функции кардиомиоцитов: 1. Одиночный генный дефект (например, дефекты в актине кардиомиоцитов при некоторых аутосомно-доминатных ДКМП; генные дефекты в хромосоме 1, 9 и 3 при других формах фамильной ДКМП). 2. Полиморфные изменения в видоизмененных генах, как в гене АПФ и других компонентов ренин-ангиотензин-альдостероновой системы (РААС), так и b2-адренергических рецепторах. 3. Малоадаптивные изменения экспрессии практически нормальных генов, которые приводят к раскодированию белков, вовлеченных в сократительную функцию сердца или структуру его полостей. Именно 3-й механизм представляется наиболее интересным как с точки зрения его вклада в развитие первичной и вторичной ДКМП, так и с позиций возможных терапевтических воздействий. Функционирование кардиомиоцитов в основе своей имеет две составлющие: 1) внутреннюю, объединяющую механизмы, ответственные за сократимость и расслабление сердца в базальных условиях, в отсутствие каких-либо внешних воздействий; 2) модулируемую, заключающую в себе те структуры, которые позволяют сердцу быстро повышать или снижать свою активность в ответ на различные физические и психоэмоциональные стимулы. Модулируемое функционирование сердца во многом определяется эндогенными биоактивными веществами, такими как нейротрансмиттеры, цитокины, аутокринные и паракринные субстанции, а также гормоны. При ХСН в пораженном миокарде происходят изменения в экспрессии генов, потенциально ответственных за обе представленные категории. В первую очередь они затрагивают контрактильные белки или их регулирующие элементы; различные механизмы, обеспечивающие соединение процессов возбуждения-сокращения; b-адренергические пути проведения; процессы, приводящие к дефициту энергетических механизмов. Вышеперечисленные аномалии нарушают работу закона Франка-Старлинга, извращают частотно-силовые взаимоотношения и ведут к развитию патологического ремоделирования сердца, полностью перестраивая его внутреннее функционирование. Описанные изменения подкрепляются нарушениями со стороны b-адренергических путей проведения, в значительной мере определяющих модулируемую функцию сердца на рецепторном и клеточном уровнях. В результате происходит значительное уменьшение количества и плотности b1-рецепторов в пораженном сердце, тогда как плотность b2-рецепторов остается практически без каких-либо изменений. В пропорциональном отношении количество b2-рецепторов возрастает до 40%, но их функционирование может быть изменено из-за нарушения в связи с аденилипциклазой. В совокупности с повышением концентрации блокирующих G-протеинов и усилением процессов b-рецепторного фосфорилирования данные изменения привносят свой вклад в нарушение сократительной функции пораженного миокарда при ХСН. Нарушение насосной функции сердца при ДКМП также может быть обусловлено и уменьшением количества самих кардиомиоцитов. На сегодняшний день известно по крайней мере два пути гибели клеток: некроз и апоптоз. Одним из типичных случаев некроза является тяжелое воспаление, сопровождаемое высвобождением продуктов клеточного распада с образованием полиморфно-клеточного инфильтрата. В отличие от некроза, апоптоз представляет собой естественный путь смерти клеток, и погибшие клетки обычно утилизируются в результате фагоцитоза без признаков какого-либо воспаления. В силу того, что кардиомиоциты являются клетками конечно-детерминированными и практически лишены способности к делению, то, естественно, их потеря в значительной мере определяет степень нарушения сократительной способности оставшегося “живого” миокарда. Происходит активация компенсаторных механизмов, основным из которых является гипертрофия оставшихся жизнеспособных кардиомиоцитов. Однако, как уже отмечалось, данные изменения носят малоадаптивный характер, сопровождаются изменением экспрессии генов и в конечном итоге ведут к дальнейшему прогрессированию нарушения сократительной функции сердца. Наряду с потерей клеточных элементов происходит активация аутоиммунных процессов с образованием антител к миозину и b1-адренорецепторам. Данные антитела чрезвычайно непросто определить, особенно при прогрессировании симптомов заболевания. Вместе с этим предполагается, что аутоиммунные процессы при кардиомиопатии активируют цитокины (фактор некроза опухоли-a (ФНО-a), интерлейкин–6), которые запускают воспалительные реакции и ведут к дальнейшему прогрессирующему поражению миокарда. Отмеченные иммунологические и воспалительные реакции не могут быть свойственны только для ДКМП и встречаются также при атеросклеротическом поражении миокарда, что отражает универсальность процессов перестройки миокарда при развитии ХСН. Если же попытаться схематически изобразить, каким образом происходит переход от левожелудочковой дисфункции к манифестированным проявлениям ХСН при ДКМП, то эта схема во многом будет универсальна и применима не только к данной патологии, как и многие из вышеописанных процессов. В этом, пожалуй, и заключается уникальность ситуации, определяющая практически равную эффективность применения ингибиторов АПФ при ХСН различной этиологии. Итак, при повреждении миокарда происходит активация различных компенсаторных механизмов с целью нормализации сердечной деятельности (табл. 2, см. рисунок). В первую очередь они затрагивают систему нейрогуморальной регуляции (НГР), что приводит к относительной стабилизации ситуации на какой-то непродолжительный отрезок времени через увеличение частоты сердечных сокращений (ЧСС) и сократительной способности миокарда, развитие гипертрофии и задержку жидкости. В случае, если причина, приведшая к поражению миокарда, не устранена или же была достаточно серьезна, длительное использование задействованных компенсаторных механизмов может приводить к постепенному их истощению. Происходит дальнейшая активизация процессов, связанных с механическим перерастяжением стенок пораженного сердца, с эффектами системы НГР и цитокинами (ФНО-a), что отражается в прогрессирующей потере контрактильных элементов (апоптоз и некроз) и развитии патологического ремоделирования кардиомиоцитов и миокарда как неотъемлемой части развития СН. Активация РААС, особенно на уровне локальных звеньев, представляет из себя одну из ключевых составляющих процессов, лежащих в основе как перестройки пораженного миокарда, так и основных симптомов ХСН. Система кининов также играет в этом не последнюю роль, чему мы имеем на сегодняшний день не одно подтверждение. Если попытаться сопоставить известные негативные эффекты основных медиаторов РААС ангиотензина II (А-II) и альдостерона и цепь основных событий, которые происходят в пораженном миокарде при ДКМП, то можно без преувеличения сказать, что применение ингибиторов АПФ у больных с данной патологией безусловно необходимо. К чему же приводит длительная чрезмерная гиперактивация РААС? • К потенцированию активности других нейро-гормональных систем, таких как симпатико-адреналовой (САС), эндотелина, также играющих важную роль в развитии апоптоза и некроза, клеточного роста и ремоделирования. • К эффектам, ведущим к ишемии почек, увеличению задержки натрия и воды и формированию отечного синдрома. • К сосудистым эффектам - коронарная и системная вазоконстрикция, провоцирующая увеличение нагрузки на сердце и потребности миокарда в кислороде. В комбинации с задержкой жидкости это ведет к гемодинамической перегрузке и перерастяжению стенки пораженного миокарда, что служит пусковым моментом к развитию апоптоза, изменению генной экспрессии и ремоделирования. • К непосредственному токсическому повреждающему действию А-II на кардиомиоциты, вызывающее их дисфункцию и некроз. • А-II является самостоятельным инициирующим фактором реакций генетического ответа и клеточного роста, что ведет к гипертрофии кардиомиоцитов и гиперплазии фибробластов (развитию фиброза). • Изменения на генетическом уровне наряду со стимуляцией оксидативного стресса и образованием свободных радикалов кислорода активируют процессы програмированной гибели клеток (апоптоза). Системе брадикинина в последние годы уделяется все большее внимание, особенно в свете той широкой дискуссии, которая развернулась в отношении преимуществ и недостатков ингибиторов АПФ в сравнении с блокаторами рецепторов А-II. Данная дискуссия стимулировала повышенный интерес к тем эффектам, которые оказывает данный агент и при ХСН. Значимость эффектов данной системы при СН превалирует над возможностью развития известных побочных влияний брадикинина. Таким образом, учитывая известный механизм действия ингибиторов АПФ, заключающийся в блокаде активности РААС и активации системы брадикинина, можно с уверенностью рекомендовать данную группу лекарственных препаратов в качестве одной из первых линий для лечения пациентов с ДКМП. Что же касается вопроса о наличии каких-либо особенностей применения ингибиторов АПФ при ДКМП в отличие от ишемической этиологии ХСН, то на сегодняшний день таковые практически отсутствуют, символизируя единый подход в лечении больных с этой тяжелой патологией: начиная от левожелудочковой дисфункции и кончая тяжелой сердечной декомпенсацией; причем чем раньше начата терапия, тем больше шансов у больного отдалить манифестацию симптомов ХСН; чем тяжелее больной, чем ниже сократительная способность его сердца, тем более значимым может быть результат. Подтверждением этому служат данные таких крупных исследований, как SOLVD и V-HeFT-II, так же как и результаты метаанализа исследований с ингибиторами АПФ (Garg R. & Yusuf S.,1995), которые выявили практически равноценное влияние данной группы лекарственных препаратов на смертность больных по причине СН. Заключение
Применение ингибиторов АПФ у больных ХСН вследствие миокардиального поражения сердца оправдано в первую очередь с патофизиологической точки зрения. Процессы апоптоза, некроза, патологического ремоделирования кардиомиоцитов и сердца, изменения экспрессии генов, коронарная и периферическая вазоконстрикция, нагрузочные условия — все эти и другие механизмы, лежащие в основе перестройки миокарда при сердечной недостаточности вследствие миокардиального поражения сердца во многом обусловлены активацией основных эффекторов РААС. Данные механизмы в основе своей универсальны и характерны в целом для поражения сердца при синдроме ХСН. Учитывая известные эффекты ингибиторов АПФ, заключающиеся не только в блокаде РААС, но и активизации системы брадикинина, препятствующей развитию гипертрофии и патологического ремоделирования сердца, применение данной группы нейрогормональных модуляторов рекомендовано с самого начала развития патологического процесса, с момента первых проявлений левожелудочковой дисфункции и прямо показано при манифестации симптомов ХСН. Список литературы Вы можете найти на сайте http://www.rmj.ru
Литература
1. Беленков Ю.Н., Мареев В.Ю., Агеев Ф.Т. Медикаментозные пути улучшения прогноза больных хронической сердечной недостаточностью, М, Инсайт, 1997.
2. J.G.F .Clealand, J.McGowan. Heart failure due to ischaemic heart disease: epidemiology, pathophysiology and progression. J Cardiovasc Pharm 1999; 33 (Suppl. 3): S17—29.
3. F.Follath. Nonischemic heart failure: epidemiology, pathophysiology and progression of disease. J Cardiovasc Pharm 1999; 33 (Suppl. 3): S31—35.
4. M.R. Bristow. Why does myocardium fail? Insights from basic science. Lancet 1998; 352 (Suppl. I): 8—14.
5. W.T. Abraham, J.D. Port, M.R. Bristow. Neurohumoral receptors in the failig heart. In P.A.Poole-Wilson, W.S.Golucci, B.M.Massie et al “Heart Failure”; Churchill Livingstone Inc. 1997: 127—41.
6. R.Garg, S.Yusuf. Overview of randomised trials of angiotensin-converting enzyme inhibitors on mortality and morbidity in patients with heart failure. JAMA 1995; 273 (18): 1450—6.
| Приложения к статье |

| Основные эффекты, связанные с повышенной активностью брадикинина:
• вазодилатация, повышение натрий- и диуреза; • блокада и регрессия процессов гипертрофии кардиомиоцитов; • блокада и регрессия процессов кардиофиброза. |
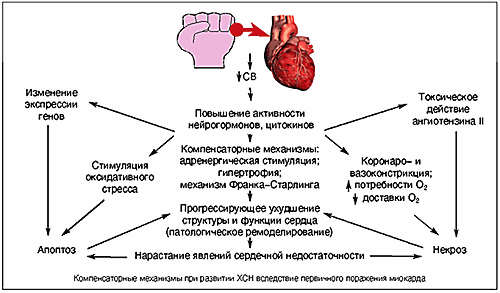
| Компенсаторные механизмы при развитии ХСН вследствие первичного поражения миокарда |




