




–Т–≤–µ–і–µ–љ–Є–µ. –Я–Њ–љ—П—В–Є–µ –Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–µ
–Т —В–µ—З–µ–љ–Є–µ –њ–Њ—Б–ї–µ–і–љ–Є—Е 10–15 –ї–µ—В –њ–Њ–љ—П—В–Є–µ «–Ї–Њ–љ—В–Є–љ—Г—Г–Љ» –њ—А–Њ—З–љ–Њ –≤–Њ—И–ї–Њ –≤ –љ–∞—Г—З–љ–Њ––Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Є–є –ї–µ–Ї—Б–Є–Ї–Њ–љ. –Я—А–Є–Љ–µ–љ–Є—В–µ–ї—М–љ–Њ –Ї –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Љ–µ–і–Є—Ж–Є–љ–µ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ (–Њ—В –∞–љ–≥–ї. continuous – –љ–µ–њ—А–µ—А—Л–≤–љ—Л–є, –њ–Њ—Б—В–Њ—П–љ–љ—Л–є) –њ–Њ–і—А–∞–Ј—Г–Љ–µ–≤–∞–µ—В –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –љ–µ–њ—А–µ—А—Л–≤–љ—Г—О —Ж–µ–њ—М —А–∞–Ј–≤–Є—В–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П – –Њ—В —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞ –і–Њ –≥–Є–±–µ–ї–Є –њ–∞—Ж–Є–µ–љ—В–∞. –Т 1991 –≥–Њ–і—Г Dzau V. –Є Braunwald E. –±—Л–ї–∞ –њ—А–µ–і–ї–Њ–ґ–µ–љ–∞ —В–µ–Њ—А–Є—П –µ–і–Є–љ–Њ–≥–Њ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞, –Ї–Њ—В–Њ—А—Г—О –≤ –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –Љ–Њ–ґ–љ–Њ —Б—З–Є—В–∞—В—М –Њ–±—Й–µ–њ—А–Є–Ј–љ–∞–љ–љ–Њ–є [1,2]. –°—Д–Њ—А–Љ—Г–ї–Є—А–Њ–≤–∞–љ–∞ –Є –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–∞ –Ї–Њ–љ—Ж–µ–њ—Ж–Є—П –њ–Њ—З–µ—З–љ–Њ–≥–Њ, –Є–ї–Є —А–µ–љ–∞–ї—М–љ–Њ–≥–Њ, –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞ –≤ —А–∞–Љ–Ї–∞—Е —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є (–°–°–Ч). –° –љ–µ–і–∞–≤–љ–µ–≥–Њ –≤—А–µ–Љ–µ–љ–Є –≤ –ї–Є—В–µ—А–∞—В—Г—А–µ —Б—В–∞–ї–Є —Г–њ–Њ—В—А–µ–±–ї—П—В—М —В–µ—А–Љ–Є–љ «–≥–Њ—А–Љ–Њ–љ–∞–ї—М–љ—Л–є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ –ґ–µ–љ—Б–Ї–Њ–≥–Њ –Ј–і–Њ—А–Њ–≤—М—П», –њ–Њ–і –Ї–Њ—В–Њ—А—Л–Љ –њ–Њ–і—А–∞–Ј—Г–Љ–µ–≤–∞—О—В –Њ—Ж–µ–љ–Ї—Г —А–∞–Ј–≤–Є—В–Є—П —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞, –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї—Г, –Љ–µ—А—Л –Є —Б—В—А–∞—В–µ–≥–Є—О –њ—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–Є –Є –ї–µ—З–µ–љ–Є—П –°–°–Ч –≤ —А–∞–Ј–ї–Є—З–љ—Л–µ –њ–µ—А–Є–Њ–і—Л –ґ–Є–Ј–љ–Є –ґ–µ–љ—Й–Є–љ—Л –≤ –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В –µ–µ –≥–Њ—А–Љ–Њ–љ–∞–ї—М–љ–Њ–≥–Њ —Б—В–∞—В—Г—Б–∞ –Є —Б–Њ—Б—В–Њ—П–љ–Є—П –њ–Њ–ї–Њ–≤–Њ–є —Б—Д–µ—А—Л [3].
–¶–µ–ї—М—О –љ–∞—Б—В–Њ—П—Й–µ–є —Б—В–∞—В—М–Є —П–≤–Є–ї–Њ—Б—М —А–∞—Б—Б–Љ–Њ—В—А–µ–љ–Є–µ —Н–≤–Њ–ї—О—Ж–Є–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П –Њ—Б–љ–Њ–≤–љ—Л—Е –Њ—А–≥–∞–љ–Њ–≤––Љ–Є—И–µ–љ–µ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є (–Р–У), —Б–µ—А–і—Ж–∞ –Є –њ–Њ—З–µ–Ї, –≤ –∞—Б–њ–µ–Ї—В–µ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Є –њ–Њ—З–µ—З–љ–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞.
–°–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ
–Є –µ–≥–Њ «–≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Є–є –Ї–∞—Б–Ї–∞–і»
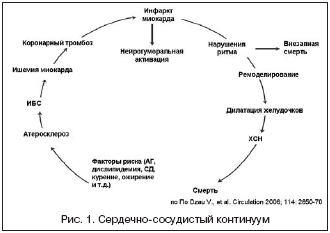
–°–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є –љ–µ–њ—А–µ—А—Л–≤–љ—Г—О —Ж–µ–њ—М —А–∞–Ј–≤–Є—В–Є—П –°–°–Ч –Њ—В —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞ –і–Њ –≥–Є–±–µ–ї–Є –њ–∞—Ж–Є–µ–љ—В–∞ (—А–Є—Б. 1). –§–∞–Ї—В–Њ—А—Л —А–Є—Б–Ї–∞, –Њ—Б–љ–Њ–≤–љ—Л–Љ–Є –Є–Ј –Ї–Њ—В–Њ—А—Л—Е —П–≤–ї—П—О—В—Б—П –Р–У, –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є—П –Є —Б–∞—Е–∞—А–љ—Л–є –і–Є–∞–±–µ—В (–°–Ф), —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В —А–∞–Ј–≤–Є—В–Є—О –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е –∞—А—В–µ—А–Є–є –Є –Є—И–µ–Љ–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є —Б–µ—А–і—Ж–∞ (–Ш–С–°). –Я–Њ—Б–ї–µ–і–љ—П—П –Љ–Њ–ґ–µ—В –Њ—Б–ї–Њ–ґ–љ—П—В—М—Б—П —А–∞–Ј–≤–Є—В–Є–µ–Љ –Є–љ—Д–∞—А–Ї—В–∞ –Љ–Є–Њ–Ї–∞—А–і–∞ (–Ш–Ь) —Б –≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є–µ–Љ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є (–•–°–Э) –Є –њ–Њ—Б–ї–µ–і—Г—О—Й–µ–є –≥–Є–±–µ–ї—М—О –њ–∞—Ж–Є–µ–љ—В–∞. –≠–≤–Њ–ї—О—Ж–Є—П —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞ –њ–Њ—Б–ї–µ –Ш–Ь –Љ–Њ–ґ–µ—В –њ—А–Њ–Є—Б—Е–Њ–і–Є—В—М –Є –њ–Њ –і—А—Г–≥–Њ–Љ—Г —Б—Ж–µ–љ–∞—А–Є—О: –љ–∞—А—Г—И–µ–љ–Є—П —Н–ї–µ–Ї—В—А–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Б–≤–Њ–є—Б—В–≤ –Љ–Є–Њ–Ї–∞—А–і–∞ –њ—А–Њ–≤–Њ—Ж–Є—А—Г—О—В –љ–∞—А—Г—И–µ–љ–Є—П —Б–µ—А–і–µ—З–љ–Њ–≥–Њ —А–Є—В–Љ–∞, –≤ —В–Њ–Љ —З–Є—Б–ї–µ –ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤—Л–µ —В–∞—Е–Є–∞—А–Є—В–Љ–Є–Є, –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–≥—Г—В —Б—В–∞—В—М –њ—А–Є—З–Є–љ–Њ–є –≤–љ–µ–Ј–∞–њ–љ–Њ–є —Б–Љ–µ—А—В–Є –±–Њ–ї—М–љ–Њ–≥–Њ [1,2,4].
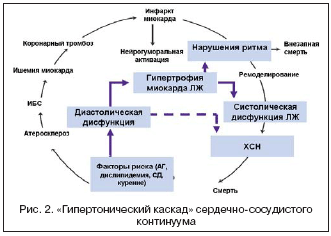
–С–Њ–ї–µ–µ —В–Њ–≥–Њ, —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ –Љ–Њ–ґ–µ—В —А–∞–Ј–≤–Є–≤–∞—В—М—Б—П –њ–Њ –±–Њ–ї–µ–µ –Ї–Њ—А–Њ—В–Ї–Њ–Љ—Г –њ—Г—В–Є, –≥–і–µ –љ–µ —Б—В–Њ–ї—М–Ї–Њ –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј, —Б–Ї–Њ–ї—М–Ї–Њ —Б–Њ–±—Б—В–≤–µ–љ–љ–Њ –Р–У –Є –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ —Б–µ—А–і—Ж–∞ –Є–≥—А–∞—О—В –Ї–ї—О—З–µ–≤—Г—О —А–Њ–ї—М. –Ґ–∞–Ї, –•–°–Э –Є —Д–∞—В–∞–ї—М–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П —А–Є—В–Љ–∞ —Г –±–Њ–ї—М–љ—Л—Е –Р–У –Љ–Њ–≥—Г—В –≤–Њ–Ј–љ–Є–Ї–∞—В—М –і–∞–ґ–µ –≤ –Њ—В—Б—Г—В—Б—В–≤–Є–µ –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е –∞—А—В–µ—А–Є–є, –Ш–С–° –Є –њ–µ—А–µ–љ–µ—Б–µ–љ–љ–Њ–≥–Њ –Ш–Ь. –Ґ–Њ –µ—Б—В—М –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ —Б–µ—А–і—Ж–∞ —Б–∞–Љ–Њ –њ–Њ —Б–µ–±–µ —П–≤–ї—П–µ—В—Б—П –і–Њ—Б—В–∞—В–Њ—З–љ—Л–Љ —Г—Б–ї–Њ–≤–Є–µ–Љ –і–Њ—Б—В–Є–ґ–µ–љ–Є—П —Д–Є–љ–∞–ї–∞ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞, –∞ –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј –Є —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б –љ–Є–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –ї–Є—И—М —Г—Б–Ї–Њ—А—П—О—В —Н—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б. –Я–Њ–і–Њ–±–љ—Л–є —Б—Ж–µ–љ–∞—А–Є–є —А–∞–Ј–≤–Є—В–Є—П —Б–Њ–±—Л—В–Є–є –Љ–Њ–ґ–µ—В –±—Л—В—М —Г—Б–ї–Њ–≤–љ–Њ –љ–∞–Ј–≤–∞–љ «–≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Є–Љ –Ї–∞—Б–Ї–∞–і–Њ–Љ» —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞ (—А–Є—Б. 2).
–Ю–і–љ–Є–Љ –Є–Ј —А–∞–љ–љ–Є—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞ –њ—А–Є –Р–У —П–≤–ї—П–µ—В—Б—П –і–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П (–Ф–Ф). –Э–µ—А–µ–і–Ї–Њ –Ф–Ф –і–∞–ґ–µ –њ—А–µ–і—И–µ—Б—В–≤—Г–µ—В —А–∞–Ј–≤–Є—В–Є—О —Б–Њ–±—Б—В–≤–µ–љ–љ–Њ –≥–Є–њ–µ—А—В—А–Њ—Д–Є–Є –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ (–У–Ы–Ц), —В–Њ –µ—Б—В—М —Г–≤–µ–ї–Є—З–µ–љ–Є—О –Љ–∞—Б—Б—Л –Љ–Є–Њ–Ї–∞—А–і–∞ [5]. –Т –Њ—Б–љ–Њ–≤–µ —А–∞–Ј–≤–Є—В–Є—П –Ф–Ф –ї–µ–ґ–∞—В –і–≤–∞ –Њ—Б–љ–Њ–≤–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞. –Я–µ—А–≤—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ – –љ–∞—А—Г—И–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ–≥–Њ —А–∞—Б—Б–ї–∞–±–ї–µ–љ–Є—П –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–Њ–≤ –Є–Љ–µ–µ—В –љ–∞–Є–±–Њ–ї—М—И–µ–µ –Ј–љ–∞—З–µ–љ–Є–µ –љ–∞ —А–∞–љ–љ–Є—Е —Н—В–∞–њ–∞—Е –Є –Њ–±—К—П—Б–љ—П–µ—В —В–Њ—В —Д–∞–Ї—В, —З—В–Њ –Ф–Ф –Њ–±—Л—З–љ–Њ –Њ–њ–µ—А–µ–ґ–∞–µ—В —А–∞–Ј–≤–Є—В–Є–µ —Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є. –Т—В–Њ—А–Њ–є –Љ–µ—Е–∞–љ–Є–Ј–Љ —Б–≤—П–Ј–∞–љ —Б–Њ —Б—В—А—Г–Ї—В—Г—А–љ–Њ–є –њ–µ—А–µ—Б—В—А–Њ–є–Ї–Њ–є –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–∞—А–љ–Њ–≥–Њ –Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤ –Љ–Є–Њ–Ї–∞—А–і–∞. –£–≤–µ–ї–Є—З–µ–љ–Є–µ –ґ–µ—Б—В–Ї–Њ—Б—В–Є –Љ–Є–Њ–Ї–∞—А–і–∞ –Ј–∞ —Б—З–µ—В —П–≤–ї–µ–љ–Є–є —Д–Є–±—А–Њ–Ј–∞ –њ—А–Є–Њ–±—А–µ—В–∞–µ—В –Њ—Б–љ–Њ–≤–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –љ–∞ –±–Њ–ї–µ–µ –њ–Њ–Ј–і–љ–Є—Е —Н—В–∞–њ–∞—Е [6–9].
–Я–Њ–Љ–Є–Љ–Њ —Н—В–Њ–≥–Њ, –љ–∞—А—Г—И–µ–љ–Є—П –і–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –Ы–Ц –Є–Љ–µ—О—В —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –≤ —А–∞–Ј–≤–Є—В–Є–Є –•–°–Э, —Б–Њ–Ї—А–∞—Й–∞—П «–њ—Г—В—М» —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞. –Ш–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –Њ–Ї–Њ–ї–Њ 50% –±–Њ–ї—М–љ—Л—Е –•–°–Э –Є–Љ–µ—О—В –Ф–Ф –њ—А–Є –љ–Њ—А–Љ–∞–ї—М–љ–Њ–є —Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –Ы–Ц [10,11].
–°–ї–µ–і—Г—О—Й–Є–Љ —Н—В–∞–њ–Њ–Љ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞ —П–≤–ї—П–µ—В—Б—П —А–∞–Ј–≤–Є—В–Є–µ —Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є. –£–Љ–µ–љ—М—И–µ–љ–Є–µ –∞–Љ–њ–ї–Є—В—Г–і—Л –Є —Б–Ї–Њ—А–Њ—Б—В–Є —Б–Њ–Ї—А–∞—Й–µ–љ–Є—П –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–Њ–≤ –Є –Љ–Є–Њ–Ї–∞—А–і–Є–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј –њ—А–Є–≤–Њ–і—П—В –Ї –љ–∞—А—Г—И–µ–љ–Є—О —Б–Њ–Ї—А–∞—В–Є—В–µ–ї—М–љ–Њ–є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є –Ы–Ц, —В–Њ –µ—Б—В—М –Ї –і–µ–Ї–Њ–Љ–њ–µ–љ—Б–∞—Ж–Є–Є –≥–Є–њ–µ—А—В—А–Њ—Д–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –Ы–Ц [6,12]. –Я–Њ –і–∞–љ–љ—Л–Љ –§—А–∞–Љ–Є–љ–≥–µ–Љ—Б–Ї–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –У–Ы–Ц, –≤—Л—П–≤–ї–µ–љ–љ–∞—П –њ—А–Є –≠–Ъ–У, —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В —А–Є—Б–Ї –•–°–Э –≤ 15 —А–∞–Ј —Г –Љ—Г–ґ—З–Є–љ –Є –≤ 12,8 —А–∞–Ј–∞ —Г –ґ–µ–љ—Й–Є–љ [13].
–Х—Й–µ –Њ–і–Є–љ «–Ї–Њ—А–Њ—В–Ї–Є–є» –њ—Г—В—М —А–∞–Ј–≤–Є—В–Є—П —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞ —Б–≤—П–Ј–∞–љ —Б –њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ —А–Є—Б–Ї–Њ–Љ —А–∞–Ј–≤–Є—В–Є—П —А–∞–Ј–љ–Њ–Њ–±—А–∞–Ј–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є —А–Є—В–Љ–∞ —Б–µ—А–і—Ж–∞ (–≤ —В–Њ–Љ —З–Є—Б–ї–µ –ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤–Њ–є —Н–Ї—Б—В—А–∞—Б–Є—Б—В–Њ–ї–Є–Є –Є —В–∞—Е–Є–Ї–∞—А–і–Є–Є), –∞ —В–∞–Ї–ґ–µ –≤–љ–µ–Ј–∞–њ–љ–Њ–є —Б–Љ–µ—А—В–Є —Г –±–Њ–ї—М–љ—Л—Е —Б –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Є–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —Б–µ—А–і—Ж–∞. –Х–µ –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М –≤–Њ–Ј—А–∞—Б—В–∞–µ—В –≤ 3 —А–∞–Ј–∞ —Г –ґ–µ–љ—Й–Є–љ –Є –≤ 6 —А–∞–Ј —Г –Љ—Г–ґ—З–Є–љ —Б –У–Ы–Ц [14].
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л–µ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Є—П –Њ —А–∞–Ј–≤–Є—В–Є–Є –°–°–Ч –њ—А–µ–і–њ–Њ–ї–∞–≥–∞—О—В –љ–µ–њ—А–µ—А—Л–≤–љ–Њ–µ —А–∞–Ј–≤–Є—В–Є–µ –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞ –Њ—В —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞ –Ї —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є—О —Б–µ—А–і—Ж–∞, —А–∞–Ј–≤–Є—В–Є—О –•–°–Э –Є, –≤ –Ї–Њ–љ–µ—З–љ–Њ–Љ –Є—В–Њ–≥–µ, –Ї –≥–Є–±–µ–ї–Є –±–Њ–ї—М–љ–Њ–≥–Њ. –Т–Њ–Ј–Љ–Њ–ґ–љ—Л –Є –±–Њ–ї–µ–µ –Ї–Њ—А–Њ—В–Ї–Є–µ –њ—Г—В–Є —Н–≤–Њ–ї—О—Ж–Є–Є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞. –Т —З–∞—Б—В–љ–Њ—Б—В–Є, –±–Њ–ї—М—И–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –Є–Љ–µ–µ—В —Б—Ж–µ–љ–∞—А–Є–є —А–∞–Ј–≤–Є—В–Є—П —Б–Њ–±—Л—В–Є–є, –Ї–ї—О—З–µ–≤—Г—О —А–Њ–ї—М –≤ –Ї–Њ—В–Њ—А–Њ–Љ –Є–≥—А–∞—О—В –Р–У –Є –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ —Б–µ—А–і—Ж–∞, –њ—А–Є–≤–Њ–і—П—Й–Є–µ –Ї —А–∞–Ј–≤–Є—В–Є—О —Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –Ы–Ц –Є —Г–≤–µ–ї–Є—З–Є–≤–∞—О—Й–Є–µ –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М –Є—И–µ–Љ–Є—З–µ—Б–Ї–Є—Е –Є –∞—А–Є—В–Љ–Є—З–µ—Б–Ї–Є—Е —Б–Њ–±—Л—В–Є–є («–≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Є–є –Ї–∞—Б–Ї–∞–і» —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞).
–Я–Њ—З–µ—З–љ—Л–є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ
–Т–Ј–∞–Є–Љ–Њ–Њ—В–љ–Њ—И–µ–љ–Є—П –Љ–µ–ґ–і—Г –Р–У –Є –њ–Њ—З–Ї–∞–Љ–Є –±–µ—Б–Ї–Њ–љ–µ—З–љ–Њ —Б–ї–Њ–ґ–љ—Л. –Я–Њ––њ—А–µ–ґ–љ–µ–Љ—Г –≤–µ–і—Г—В—Б—П –і–Є—Б–Ї—Г—Б—Б–Є–Є –њ–Њ –њ–Њ–≤–Њ–і—Г —В–Њ–≥–Њ, —П–≤–ї—П–µ—В—Б—П –ї–Є –љ–µ—Д—А–Њ–љ –њ—А–Є—З–Є–љ–Њ–є, «–ґ–µ—А—В–≤–Њ–є» –Є/–Є–ї–Є «—Б–Њ—Г—З–∞—Б—В–љ–Є–Ї–Њ–Љ» –≤ —А–∞–Ј–≤–Є—В–Є–Є –Р–У.
–Я–Њ—З–Ї–Є –Љ–Њ–≥—Г—В –Є–Љ–µ—В—М –њ–µ—А–≤–Є—З–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –≤ —А–∞–Ј–≤–Є—В–Є–Є –Р–У – –њ–Њ—Б—В—Г–ї–∞—В, –Ї–Њ—В–Њ—А—Л–є –≤–њ–µ—А–≤—Л–µ –≤—Л–і–≤–Є–љ—Г–ї–Є Guyton et al. –Э–µ—Б–Ї–Њ–ї—М–Ї–Њ —В–µ–Њ—А–Є–є –Њ–±—К—П—Б–љ—П—О—В «–њ–µ—А–≤–Є—З–љ–Њ—Б—В—М» –њ–Њ—З–µ—З–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –≤ –≥–µ–љ–µ–Ј–µ –Р–У: –і–µ—Д–µ–Ї—В—Л –Љ–µ–Љ–±—А–∞–љ—Л –њ–Њ—З–µ—З–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–µ–≤, –≤—А–Њ–ґ–і–µ–љ–љ–Њ–µ —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –љ–µ—Д—А–Њ–љ–Њ–≤ (–Њ–ї–Є–≥–Њ–љ–µ—Д—А–Њ–љ–Є—П) –Є–ї–Є —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–≥–Њ —Д–Є–ї—М—В—А–∞—Ж–Є–Њ–љ–љ–Њ–≥–Њ —А–µ–Ј–µ—А–≤–∞ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤, –≥–µ—В–µ—А–Њ–≥–µ–љ–љ–Њ—Б—В—М –њ–Њ–њ—Г–ї—П—Ж–Є–Є –љ–µ—Д—А–Њ–љ–Њ–≤ (–Є—И–µ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л–µ –Є –љ–µ–Є—И–µ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л–µ), –≤—А–Њ–ґ–і–µ–љ–љ–∞—П –≥–Є–њ–µ—А—З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В—М —Н—Д—Д–µ—А–µ–љ—В–љ–Њ–є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –∞—А—В–µ—А–Є–Њ–ї—Л –Ї –њ—А–µ—Б—Б–Њ—А–љ—Л–Љ —Б—В–Є–Љ—Г–ї–∞–Љ, –≤ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –Ї –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ—Г II (–Р–ҐII). –Т —Ж–µ–ї–Њ–Љ –≤—Б–µ —Н—В–Є –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –≤–µ–і—Г—В –Ї –љ–∞—А—Г—И–µ–љ–Є—О —А–∞–≤–љ–Њ–≤–µ—Б–Є—П –Љ–µ–ґ–і—Г –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є–µ–Љ –Є –≤—Л–≤–µ–і–µ–љ–Є–µ–Љ –ґ–Є–і–Ї–Њ—Б—В–Є –≤ —Б—В–Њ—А–Њ–љ—Г –µ–µ –Ј–∞–і–µ—А–ґ–Ї–Є –≤ –Њ—А–≥–∞–љ–Є–Ј–Љ–µ [15,16].
–° –і—А—Г–≥–Њ–є —Б—В–Њ—А–Њ–љ—Л, –њ–Њ—З–Ї–Є —П–≤–ї—П—О—В—Б—П –Њ–і–љ–Є–Љ –Є–Ј –Њ—Б–љ–Њ–≤–љ—Л—Е –Њ—А–≥–∞–љ–Њ–≤––Љ–Є—И–µ–љ–µ–є –Р–У. –Я–Њ –і–∞–љ–љ—Л–Љ USRDS (United States Renal Data System), –Р–У —П–≤–ї—П–µ—В—Б—П –≤—В–Њ—А—Л–Љ –њ–Њ –Ј–љ–∞—З–Є–Љ–Њ—Б—В–Є (–њ–Њ—Б–ї–µ –°–Ф) —Д–∞–Ї—В–Њ—А–Њ–Љ —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ—З–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є (–•–Я–Э) [17].
–Ш–Ј–Љ–µ–љ–µ–љ–Є—П, –њ—А–Њ–Є—Б—Е–Њ–і—П—Й–Є–µ –≤ –њ–Њ—З–Ї–∞—Е, –Љ–Њ–ґ–љ–Њ –њ—А–µ–і—Б—В–∞–≤–Є—В—М –≤ –≤–Є–і–µ –љ–µ–њ—А–µ—А—Л–≤–љ–Њ–є —Ж–µ–њ–Є – –њ–Њ—З–µ—З–љ–Њ–≥–Њ, –Є–ї–Є —А–µ–љ–∞–ї—М–љ–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞ – –Њ—В —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞ (–Р–У, –°–Ф –Є –і—А.) –і–Њ —А–∞–Ј–≤–Є—В–Є—П —В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є –•–Я–Э –Є –≥–Є–±–µ–ї–Є –±–Њ–ї—М–љ–Њ–≥–Њ (—А–Є—Б. 3).
–Ю–і–љ–Є–Љ –Є–Ј –љ–∞–Є–±–Њ–ї–µ–µ —А–∞–љ–љ–Є—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–Њ—З–µ–Ї —П–≤–ї—П—О—В—Б—П —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П –њ–Њ—З–µ—З–љ–Њ–є –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є–Ї–Є —Б —А–∞–Ј–≤–Є—В–Є–µ–Љ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –≥–Є–њ–µ—А–њ–µ—А—Д—Г–Ј–Є–Є –Є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є. –Т–љ—Г—В—А–Є–Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–µ –і–∞–≤–ї–µ–љ–Є–µ –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П –і–≤—Г–Љ—П –Њ—Б–љ–Њ–≤–љ—Л–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є – —Г—А–Њ–≤–љ–µ–Љ —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –Р–Ф –Є —Б–Њ–Њ—В–љ–Њ—И–µ–љ–Є–µ–Љ –Љ–µ–ґ–і—Г —В–Њ–љ—Г—Б–Њ–Љ –∞—Д—Д–µ—А–µ–љ—В–љ–Њ–є –Є —Н—Д—Д–µ—А–µ–љ—В–љ–Њ–є –∞—А—В–µ—А–Є–Њ–ї. –Т –љ–Њ—А–Љ–µ –њ—А–Є –њ–Њ–≤—Л—И–µ–љ–Є–Є —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –Р–Ф –њ—А–Њ–Є—Б—Е–Њ–і–Є—В —Б—Г–ґ–µ–љ–Є–µ –∞—Д—Д–µ—А–µ–љ—В–љ–Њ–є –Є —А–∞—Б—И–Є—А–µ–љ–Є–µ —Н—Д—Д–µ—А–µ–љ—В–љ–Њ–є –∞—А—В–µ—А–Є–Њ–ї—Л, —З—В–Њ –њ—А–µ–њ—П—В—Б—В–≤—Г–µ—В –њ–µ—А–µ–і–∞—З–µ –њ–Њ–≤—Л—И–µ–љ–љ–Њ–≥–Њ –Р–Ф –љ–∞ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤—Л–µ –Ї–∞–њ–Є–ї–ї—П—А—Л. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —А–∞–Ј–≤–Є—В–Є–µ –≥–Є–њ–µ—А–њ–µ—А—Д—Г–Ј–Є–Є –Є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ –њ—А–Є 1) —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–Љ –њ–Њ–≤—Л—И–µ–љ–Є–Є —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –Р–Ф –Є 2) –љ–∞—А—Г—И–µ–љ–Є–Є –∞—Г—В–Њ—А–µ–≥—Г–ї—П—Ж–Є–Є —В–Њ–љ—Г—Б–∞ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤—Л—Е –∞—А—В–µ—А–Є–Њ–ї. –Т —Н—В–Є—Е —Г—Б–ї–Њ–≤–Є—П—Е –і–Є–ї–∞—В–∞—Ж–Є—П –∞—Д—Д–µ—А–µ–љ—В–љ–Њ–є –∞—А—В–µ—А–Є–Њ–ї—Л —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –њ–µ—А–µ–і–∞—З–µ –њ–Њ–≤—Л—И–µ–љ–љ–Њ–≥–Њ –Р–Ф –љ–∞ –Ї–ї—Г–±–Њ—З–Ї–Є [16,18–22].
–Ф–ї–Є—В–µ–ї—М–љ–Њ–µ —Б—Г—Й–µ—Б—В–≤–Њ–≤–∞–љ–Є–µ –≥–Є–њ–µ—А–њ–µ—А—Д—Г–Ј–Є–Є –Є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —А–∞–Ј–≤–Є—В–Є—О —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є, –Ї–Њ—В–Њ—А—Л–µ –Ј–∞—В—А–∞–≥–Є–≤–∞—О—В –њ–Њ—З–µ—З–љ—Л–µ –Ї–ї—Г–±–Њ—З–Ї–Є (–Ї–∞–њ–Є–ї–ї—П—А—Л, –±–∞–Ј–∞–ї—М–љ—Г—О –Љ–µ–Љ–±—А–∞–љ—Г, –Љ–µ–Ј–∞–љ–≥–Є–є), –∞ —В–∞–Ї–ґ–µ –њ–Њ—З–µ—З–љ—Л–µ —Б–Њ—Б—Г–і—Л. –Э–∞–Є–±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ–љ—Л–µ —Б—В—А—Г–Ї—В—Г—А–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤–Њ–Ј–љ–Є–Ї–∞—О—В –≤ –∞—Д—Д–µ—А–µ–љ—В–љ–Њ–є –∞—А—В–µ—А–Є–Њ–ї–µ, –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–Њ –њ–Њ–і–≤–µ—А–ґ–µ–љ–љ–Њ–є –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—О –њ–Њ–≤—Л—И–µ–љ–љ–Њ–≥–Њ –Р–Ф. –Э–µ—Д—А–Њ–∞–љ–≥–Є–Њ—Б–Ї–ї–µ—А–Њ–Ј –њ—А–Є–≤–Њ–і–Є—В –Ї —Г–Љ–µ–љ—М—И–µ–љ–Є—О –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–≥–Њ –Ї—А–Њ–≤–Њ—В–Њ–Ї–∞ –Є –Є—И–µ–Љ–Є–Є, –∞ —В–∞–Ї–ґ–µ –Ї –≥–Є–±–µ–ї–Є —З–∞—Б—В–Є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤. –Ш–Љ–µ–љ–љ–Њ —Н—В–Њ—В –Љ–µ—Е–∞–љ–Є–Ј–Љ –љ–∞—А—Г—И–µ–љ–Є—П –њ–Њ—З–µ—З–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –љ–∞ —Д–Њ–љ–µ –Р–У –±—Л–ї –Њ–њ–Є—Б–∞–љ Volhard –Є Fahr [19]. –Я—А–Є –Ї—А–Є—В–Є—З–µ—Б–Ї–Њ–Љ —Г–Љ–µ–љ—М—И–µ–љ–Є–Є –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ —Д—Г–љ–Ї—Ж–Є–Њ–љ–Є—А—Г—О—Й–Є—Е –Ї–ї—Г–±–Њ—З–Ї–Њ–≤ —А–∞–Ј–≤–Є–≤–∞—О—В—Б—П –∞–і–∞–њ—В–Є–≤–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤ –Њ—Б—В–∞–≤—И–Є—Е—Б—П –љ–µ—Д—А–Њ–љ–∞—Е —Б –і–Є–ї–∞—В–∞—Ж–Є–µ–є –∞—Д—Д–µ—А–µ–љ—В–љ–Њ–є –∞—А—В–µ—А–Є–Њ–ї—Л, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л–µ –љ–∞ –њ–Њ–і–і–µ—А–ґ–∞–љ–Є–µ —Н–Ї—Б–Ї—А–µ—В–Њ—А–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї. –Ю–і–љ–∞–Ї–Њ —Б–Њ –≤—А–µ–Љ–µ–љ–µ–Љ —Н—В–Њ—В –∞–і–∞–њ—В–Є–≤–љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ —Б—В–∞–љ–Њ–≤–Є—В—Б—П —Д–∞–Ї—В–Њ—А–Њ–Љ –њ—А–Њ–≥—А–µ—Б—Б–Є–Є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є. –Я–µ—А–µ–і–∞—З–∞ –њ–Њ–≤—Л—И–µ–љ–љ–Њ–≥–Њ –Р–Ф –љ–∞ –Є–љ—В–∞–Ї—В–љ—Л–µ –Ї–ї—Г–±–Њ—З–Ї–Є, —А–∞–Ј–≤–Є—В–Є–µ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –≥–Є–њ–µ—А–њ–µ—А—Д—Г–Ј–Є–Є –Є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є –њ—А–Є–≤–Њ–і—П—В –Ї —Б—В—А—Г–Ї—В—Г—А–љ—Л–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ, –і–∞–ї—М–љ–µ–є—И–µ–Љ—Г —Б–љ–Є–ґ–µ–љ–Є—О –њ–Њ—З–µ—З–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є —Б —А–∞–Ј–≤–Є—В–Є–µ–Љ —В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є –•–Я–Э –Є –≥–Є–±–µ–ї—М—О –±–Њ–ї—М–љ–Њ–≥–Њ [16, 18–22].
–Я–Њ—З–µ—З–љ—Л–є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ –Љ–Њ–ґ–µ—В –±—Л—В—М –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ –≤ –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ –Є–љ–Њ–Љ –≤–Є–і–µ – —Б —В–Њ—З–Ї–Є –Ј—А–µ–љ–Є—П –Ї–ї–Є–љ–Є–Ї–Њ––ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л—Е –Љ–∞—А–Ї–µ—А–Њ–≤ –љ–µ—Д—А–Њ–њ–∞—В–Є–Є, –Ї–Њ—В–Њ—А—Л–µ –≤–Њ –Љ–љ–Њ–≥–Њ–Љ –Њ—В—А–∞–ґ–∞—О—В –Њ–њ–Є—Б–∞–љ–љ—Л–µ –≤—Л—И–µ –њ–∞—В–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л. –Э–∞–Є–±–Њ–ї–µ–µ —А–∞–љ–љ–Є–Љ («—Б—Г–±–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ») –µ–µ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ —П–≤–ї—П–µ—В—Б—П –Љ–Є–Ї—А–Њ–∞–ї—М–±—Г–Љ–Є–љ—Г—А–Є—П (–Ь–Є–Р), –Ї–Њ—В–Њ—А–∞—П –Љ–Њ–ґ–µ—В –±—Л—В—М –≤—Л—П–≤–ї–µ–љ–∞ —Г 5–30% –±–Њ–ї—М–љ—Л—Е –Р–У [23,24]. –Х—Б–ї–Є —Г—А–Њ–≤–µ–љ—М —Н–Ї—Б–Ї—А–µ—Ж–Є–Є –±–µ–ї–Ї–∞ –њ—А–µ–≤—Л—И–∞–µ—В 300 –Љ–≥/—Б—Г—В., –≥–Њ–≤–Њ—А—П—В –Њ–± –∞–ї—М–±—Г–Љ–Є–љ—Г—А–Є–Є, –Є–ї–Є –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є. –£–≤–µ–ї–Є—З–µ–љ–Є–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Ї—А–µ–∞—В–Є–љ–Є–љ–∞ –≤ —Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є (>1,5 –Љ–≥/–і–ї —Г –Љ—Г–ґ—З–Є–љ –Є >1,4 –Љ–≥/–і–ї —Г –ґ–µ–љ—Й–Є–љ) –Є–ї–Є —Б–љ–Є–ґ–µ–љ–Є–µ —Б–Ї–Њ—А–Њ—Б—В–Є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є —Д–Є–ї—М—В—А–∞—Ж–Є–Є (<60–70 –Љ–ї/–Љ–Є–љ.) –њ–Њ–Ј–≤–Њ–ї—П–µ—В –і–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞—В—М –љ–∞—З–∞–ї—М–љ—Г—О –њ–Њ—З–µ—З–љ—Г—О –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М [25]. –Э–∞–Ї–Њ–љ–µ—Ж, –њ–Њ—Б–ї–µ–і–љ–µ–є —Б—В–∞–і–Є–µ–є –њ–Њ—З–µ—З–љ–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞ —П–≤–ї—П–µ—В—Б—П —В–µ—А–Љ–Є–љ–∞–ї—М–љ–∞—П –•–Я–Э.
–Я–∞—В–Њ–Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—П –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ
–њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞
–Я—А–Њ—Ж–µ—Б—Б—Л —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є—П —Б–µ—А–і—Ж–∞ –Ј–∞—В—А–∞–≥–Є–≤–∞—О—В –≤—Б–µ –µ–≥–Њ –Ї–ї–µ—В–Њ—З–љ—Л–µ –Є –љ–µ–Ї–ї–µ—В–Њ—З–љ—Л–µ —Н–ї–µ–Љ–µ–љ—В—Л: –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В—Л, –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ—Л–є –Љ–∞—В—А–Є–Ї—Б, —Д–Є–±—А–Њ–±–ї–∞—Б—В—Л, –Љ–∞–Ї—А–Њ—Д–∞–≥–Є, —В—Г—З–љ—Л–µ –Ї–ї–µ—В–Ї–Є –Є –Ї–Њ—А–Њ–љ–∞—А–љ—Л–µ —Б–Њ—Б—Г–і—Л (—А–Є—Б. 4) [7].
–Ш–Ј–Љ–µ–љ–µ–љ–Є—П –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–Њ–≤ –≤–Ї–ї—О—З–∞—О—В —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –і–Є–∞–Љ–µ—В—А–∞ –Є –і–ї–Є–љ—Л –Ї–ї–µ—В–Њ–Ї. –Ш—Е —З–Є—Б–ї–Њ –Є –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –Ї–ї–µ—В–Њ—З–љ—Л—Е —Б–ї–Њ–µ–≤, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –љ–µ –Є–Ј–Љ–µ–љ—П—О—В—Б—П. –Я—А–Є –њ–µ—А–µ–≥—А—Г–Ј–Ї–µ –і–∞–≤–ї–µ–љ–Є–µ–Љ –љ–∞–Є–±–Њ–ї–µ–µ –Њ—В—З–µ—В–ї–Є–≤–Њ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —И–Є—А–Є–љ—Л (–і–Є–∞–Љ–µ—В—А–∞) –Ї–ї–µ—В–Њ–Ї (–Ї–Њ–љ—Ж–µ–љ—В—А–Є—З–µ—Б–Ї–∞—П –У–Ы–Ц), —В–Њ–≥–і–∞ –Ї–∞–Ї –њ—А–Є –њ–µ—А–µ–≥—А—Г–Ј–Ї–µ –Њ–±—К–µ–Љ–Њ–Љ –Њ—В–Љ–µ—З–∞–µ—В—Б—П –≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ —Г–і–ї–Є–љ–µ–љ–Є–µ –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–Њ–≤ (—Н–Ї—Б—Ж–µ–љ—В—А–Є—З–µ—Б–Ї–∞—П –У–Ы–Ц). –£–≤–µ–ї–Є—З–µ–љ–Є–µ —А–∞–Ј–Љ–µ—А–Њ–≤ –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–Њ–≤ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –Ј–∞ —Б—З–µ—В –∞–Ї—В–Є–≤–Є–Ј–∞—Ж–Є–Є —Б–Є–љ—В–µ—В–Є—З–µ—Б–Ї–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –Ї–ї–µ—В–Њ–Ї –Є –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—П –±–µ–ї–Ї–∞, –∞ —В–∞–Ї–ґ–µ —Г–≤–µ–ї–Є—З–µ–љ–Є—П —З–Є—Б–ї–∞ –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ—Л—Е –Њ—А–≥–∞–љ–µ–ї–ї (—А–Є–±–Њ—Б–Њ–Љ, –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –Є –і—А.). –£–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –Є —Б–Њ–і–µ—А–ґ–∞–љ–Є–µ –Ф–Э–Ъ –≤ –Ї–ї–µ—В–Ї–∞—Е. –Э–∞—А—П–і—Г —Б –њ—А–Њ—Ж–µ—Б—Б–∞–Љ–Є –≥–Є–њ–µ—А—В—А–Њ—Д–Є–Є –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–Њ–≤ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –Є—Е –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–∞—П –≥–Є–±–µ–ї—М –≤—Б–ї–µ–і—Б—В–≤–Є–µ –∞–њ–Њ–њ—В–Њ–Ј–∞, –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —З–Є—Б–ї–Њ –Љ—Л—И–µ—З–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –Љ–Є–Њ–Ї–∞—А–і–∞ –Љ–Њ–ґ–µ—В –љ–µ–Њ–±—А–∞—В–Є–Љ–Њ —Г–Љ–µ–љ—М—И–∞—В—М—Б—П [26].
–°—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Ј–∞—В—А–∞–≥–Є–≤–∞—О—В –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ—Л–є –Љ–∞—В—А–Є–Ї—Б, –Њ—Б–љ–Њ–≤–љ—Л–Љ–Є —Б–Њ—Б—В–∞–≤–ї—П—О—Й–Є–Љ–Є –Ї–Њ—В–Њ—А–Њ–≥–Њ —П–≤–ї—П—О—В—Б—П –Ї–Њ–ї–ї–∞–≥–µ–љ I –Є III —В–Є–њ–∞, —Д–Є–±—А–Њ–љ–µ–Ї—В–Є–љ –Є –ї–∞–Љ–Є–љ–Є–љ. –У–Є–њ–µ—А—В—А–Њ—Д–Є—П –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–Њ–≤ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –Ј–∞–Ї–Њ–љ–Њ–Љ–µ—А–љ—Л–Љ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ –Њ–±—Й–µ–≥–Њ —Б–Њ–і–µ—А–ґ–∞–љ–Є—П –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ—Л—Е –±–µ–ї–Ї–Њ–≤, –љ–Њ –Є—Е –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П (–Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ –Љ–∞—Б—Б—Л —В–Ї–∞–љ–Є) –Њ—Б—В–∞–µ—В—Б—П –љ–µ–Є–Ј–Љ–µ–љ–љ–Њ–є. –Т —В–Њ–Љ —Б–ї—Г—З–∞–µ, –Ї–Њ–≥–і–∞ —Б–Њ—Е—А–∞–љ—П–µ—В—Б—П –љ–Њ—А–Љ–∞–ї—М–љ—Л–є —В–Ї–∞–љ–µ–≤–Њ–є —Б–Њ—Б—В–∞–≤ –Ы–Ц, –≥–Є–њ–µ—А—В—А–Њ—Д–Є—П —П–≤–ї—П–µ—В—Б—П —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є, –Є–ї–Є –∞–і–∞–њ—В–Є–≤–љ–Њ–є. –Я—А–Є –Р–У –њ—А–Њ–Є—Б—Е–Њ–і–Є—В —Б–і–≤–Є–≥ –і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Њ–≥–Њ —А–∞–≤–љ–Њ–≤–µ—Б–Є—П –Љ–µ–ґ–і—Г –њ—А–Њ—Ж–µ—Б—Б–∞–Љ–Є —Б–Є–љ—В–µ–Ј–∞ –Є –і–µ–≥—А–∞–і–∞—Ж–Є–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–∞, –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —З–µ–≥–Њ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –і–Њ–ї—П –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞, —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Д–Є–±—А–Њ–Ј. –Ш–Љ–µ–љ–љ–Њ —Д–Є–±—А–Њ–Ј —П–≤–ї—П–µ—В—Б—П –Њ—В–ї–Є—З–Є—В–µ–ї—М–љ–Њ–є —З–µ—А—В–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –і–µ–Ј–∞–і–∞–њ—В–Є–≤–љ–Њ–є –≥–Є–њ–µ—А—В—А–Њ—Д–Є–Є. –Ф–ї—П –У–Ы–Ц –љ–∞ —Д–Њ–љ–µ –Р–У —Е–∞—А–∞–Ї—В–µ—А–µ–љ —А–µ–∞–Ї—В–Є–≤–љ—Л–є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–є –Є –њ–µ—А–Є–≤–∞—Б–Ї—Г–ї—П—А–љ—Л–є —Д–Є–±—А–Њ–Ј [27].
–Т –њ—А–Њ—Ж–µ—Б—Б–µ —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—П –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–µ—А–і—Ж–∞ —Б—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ—А–µ—В–µ—А–њ–µ–≤–∞—О—В –Ї–Њ—А–Њ–љ–∞—А–љ—Л–µ —Б–Њ—Б—Г–і—Л – –Ї–∞–Ї –Ї—А—Г–њ–љ—Л–µ –∞—А—В–µ—А–Є–Є, —В–∞–Ї –Є –Љ–µ–ї–Ї–Є–µ –Є–љ—В—А–∞–Љ—Г—А–∞–ї—М–љ—Л–µ —Б–Њ—Б—Г–і—Л. –Т —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–љ–Є–ґ–µ–љ–Є—П —А–µ–Ј–µ—А–≤–∞ –Ї–Њ—А–Њ–љ–∞—А–љ–Њ–≥–Њ –Ї—А–Њ–≤–Њ—В–Њ–Ї–∞ –У–Ы–Ц –Љ–Њ–ґ–µ—В —Б–Њ—З–µ—В–∞—В—М—Б—П —Б –Є—И–µ–Љ–Є–µ–є –Љ–Є–Њ–Ї–∞—А–і–∞ –і–∞–ґ–µ –≤ –Њ—В—Б—Г—В—Б—В–≤–Є–µ –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е –∞—А—В–µ—А–Є–є. –Ю—Б–љ–Њ–≤–љ—Л–Љ–Є –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞–Љ–Є —Н—В–Њ–≥–Њ —П–≤–ї–µ–љ–Є—П —П–≤–ї—П—О—В—Б—П [28–30]:
• —Б–і–∞–≤–ї–µ–љ–Є–µ –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е —Б–Њ—Б—Г–і–Њ–≤ –Є–Ј–≤–љ–µ (–њ–Њ–≤—Л—И–µ–љ–Є–µ –Ї–Њ–љ–µ—З–љ–Њ––і–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –Љ–∞—Б—Б—Л –Ы–Ц, –Љ–Є–Њ–Ї–∞—А–і–Є–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј);
• —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–µ –љ–∞—А—Г—И–µ–љ–Є–µ —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е —Б–Њ—Б—Г–і–Њ–≤ –Ї –і–Є–ї–∞—В–∞—Ж–Є–Є;
• —Б—В—А—Г–Ї—В—Г—А–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Ї—А—Г–њ–љ—Л—Е –Є –Љ–µ–ї–Ї–Є—Е –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е —Б–Њ—Б—Г–і–Њ–≤.
–Т –Ї—А—Г–њ–љ—Л—Е –∞—А—В–µ—А–Є—П—Е –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є—П —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Є –≥–ї–∞–і–Ї–Њ–Љ—Л—И–µ—З–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –Є–љ—В–Є–Љ—Л, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –Љ–Њ–љ–Њ—Ж–Є—В–Њ–≤ –Є –Є–Ј–±—Л—В–Њ—З–љ–Њ–µ –Њ—В–ї–Њ–ґ–µ–љ–Є–µ –±–µ–ї–Ї–Њ–≤ –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞. –Т —Б—А–µ–і–љ–µ–є –Њ–±–Њ–ї–Њ—З–Ї–µ (–Љ–µ–і–Є–Є) –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –≥–Є–њ–µ—А–њ–ї–∞–Ј–Є—П –Є/–Є–ї–Є –≥–Є–њ–µ—А—В—А–Њ—Д–Є—П –≥–ї–∞–і–Ї–Њ–Љ—Л—И–µ—З–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –Є —Д–Є–±—А–Њ–Ј. –Ч–∞ —Б—З–µ—В —Г–≤–µ–ї–Є—З–µ–љ–Є—П —В–Њ–ї—Й–Є–љ—Л —Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б—В–µ–љ–Ї–Є –љ–∞—А—Г—И–∞–µ—В—Б—П –µ–µ —А–∞—Б—В—П–ґ–Є–Љ–Њ—Б—В—М –Є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –Ї –і–Є–ї–∞—В–∞—Ж–Є–Є –≤ –Њ—В–≤–µ—В –љ–∞ –љ–µ–є—А–Њ–≥—Г–Љ–Њ—А–∞–ї—М–љ—Л–µ —Б—В–Є–Љ—Г–ї—Л. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —Б–Њ–Њ—В–љ–Њ—И–µ–љ–Є—П —Б—В–µ–љ–Ї–Є –Ї –њ—А–Њ—Б–≤–µ—В—Г —Б–Њ—Б—Г–і–∞ —Б–∞–Љ–Њ –њ–Њ —Б–µ–±–µ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —А–∞–Ј–≤–Є—В–Є—О –Є—И–µ–Љ–Є–Є –Љ–Є–Њ–Ї–∞—А–і–∞ [28,31].
–Ш–Ј–Љ–µ–љ–µ–љ–Є—П —Б–µ—А–і—Ж–∞ –њ—А–Є –Р–У —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г—О—В—Б—П —Г–Љ–µ–љ—М—И–µ–љ–Є–µ–Љ –њ–ї–Њ—В–љ–Њ—Б—В–Є –Љ–Є–Ї—А–Њ—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–µ—В–Є –Љ–Є–Њ–Ї–∞—А–і–∞ (—А–∞—А–µ—Д–Є–Ї–∞—Ж–Є—П), –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —З–µ–≥–Њ —Г—Е—Г–і—И–∞–µ—В—Б—П –Ї—А–Њ–≤–Њ—Б–љ–∞–±–ґ–µ–љ–Є–µ —Б–µ—А–і–µ—З–љ–Њ–є –Љ—Л—И—Ж—Л –Є —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –і–Є—Д—Д—Г–Ј–Є–Њ–љ–љ–Њ–µ —А–∞—Б—Б—В–Њ—П–љ–Є–µ –Љ–µ–ґ–і—Г –Ї–∞–њ–Є–ї–ї—П—А–∞–Љ–Є –Є –Ї–ї–µ—В–Ї–∞–Љ–Є. –Ю—Б–љ–Њ–≤–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ —А–∞—А–µ—Д–Є–Ї–∞—Ж–Є—П –њ—А–Є–Њ–±—А–µ—В–∞–µ—В —Г –њ–Њ–ґ–Є–ї—Л—Е –±–Њ–ї—М–љ—Л—Е, –Ї–Њ–≥–і–∞ —В–µ–Љ–њ—Л —Г–≤–µ–ї–Є—З–µ–љ–Є—П –Љ–∞—Б—Б—Л –Љ–Є–Њ–Ї–∞—А–і–∞ –њ—А–µ–≤–Њ—Б—Е–Њ–і—П—В —Б–Ї–Њ—А–Њ—Б—В—М –љ–µ–Њ–≤–∞—Б–Ї—Г–ї—П—А–Є–Ј–∞—Ж–Є–Є [28,31].
–Я–∞—В–Њ–Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—П –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ
–њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–Њ—З–µ–Ї
–Т –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –њ—А–∞–Ї—В–Є–Ї–µ –і–Є–∞–≥–љ–Њ–Ј –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П (–љ–µ—Д—А–Њ—Б–Ї–ї–µ—А–Њ–Ј, –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–≤–љ–∞—П –љ–µ—Д—А–Њ–њ–∞—В–Є—П) –Њ–±—Л—З–љ–Њ —Б—В–∞–≤—П—В –љ–∞ –Њ—Б–љ–Њ–≤–∞–љ–Є–Є –љ–∞–ї–Є—З–Є—П –Р–У, –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –њ–Њ—А–∞–ґ–µ–љ–Є—П –і—А—Г–≥–Є—Е –Њ—А–≥–∞–љ–Њ–≤––Љ–Є—И–µ–љ–µ–є, –∞ —В–∞–Ї–ґ–µ –ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л—Е –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї. –Ь–µ–ґ–і—Г —В–µ–Љ, –њ–Њ –і–∞–љ–љ—Л–Љ –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є —Б –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ–Љ –±–Є–Њ–њ—Б–Є–Є, –±—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–є –і–Є–∞–≥–љ–Њ–Ј –љ–µ—Д—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В—Б—П –ї–Є—И—М –≤ 40–75% —Б–ї—Г—З–∞–µ–≤ [32,33]. –Э–∞ –Њ—Б–љ–Њ–≤–∞–љ–Є–Є —Н—В–Њ–≥–Њ –Љ–љ–Њ–≥–Є–µ –∞–≤—В–Њ—А—Л —Б—З–Є—В–∞—О—В, —З—В–Њ –Њ –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–Є –њ–Њ—З–µ–Ї –Љ–Њ–ґ–љ–Њ –≥–Њ–≤–Њ—А–Є—В—М —В–Њ–ї—М–Ї–Њ –њ–Њ—Б–ї–µ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є—П –і–Є–∞–≥–љ–Њ–Ј–∞. –°–ї–µ–і—Г–µ—В, –Њ–і–љ–∞–Ї–Њ, –Ј–∞–Љ–µ—В–Є—В—М, —З—В–Њ –≤ –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –і–Њ—Б—В–Њ–≤–µ—А–љ—Л—Е –і–∞–љ–љ—Л—Е –њ–Њ –≥–Є—Б—В–Њ–њ–∞—В–Њ–ї–Њ–≥–Є–Є –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ—З–Ї–Є, –≤ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –µ–µ –љ–∞—З–∞–ї—М–љ—Л–Љ–Є –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є –Є –Љ—П–≥–Ї–Њ–є –•–Я–Э, –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ.
–Ш–Ј–Љ–µ–љ–µ–љ–Є—П –њ–Њ—З–µ–Ї –љ–∞ —Д–Њ–љ–µ –Р–У –Ј–∞—В—А–∞–≥–Є–≤–∞—О—В –Ї–ї—Г–±–Њ—З–Ї–Є —Б –Є—Е –Ї–∞–њ–Є–ї–ї—П—А–∞–Љ–Є, –±–∞–Ј–∞–ї—М–љ–Њ–є –Љ–µ–Љ–±—А–∞–љ–Њ–є –Є –Љ–µ–Ј–∞–љ–≥–Є–µ–Љ (–≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј), –Ї—А—Г–њ–љ—Л–µ –Є –Љ–µ–ї–Ї–Є–µ —Б–Њ—Б—Г–і—Л (–∞—А—В–µ—А–Є–Њ—Б–Ї–ї–µ—А–Њ–Ј, –∞—А—В–µ—А–Є–Њ–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј), –∞ —В–∞–Ї–ґ–µ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–є (–љ–µ—Д—А–Њ—Б–Ї–ї–µ—А–Њ–Ј –≤ «–Є—Б—В–Њ—А–Є—З–µ—Б–Ї–Њ–Љ» –Ј–љ–∞—З–µ–љ–Є–Є —Н—В–Њ–≥–Њ —Б–ї–Њ–≤–∞) (—А–Є—Б. 4) [18,34,35].
–•–∞—А–∞–Ї—В–µ—А–љ–Њ —Д–Њ–Ї–∞–ї—М–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤, –≤–Њ–Ј–љ–Є–Ї–∞–µ—В –≥–µ—В–µ—А–Њ–≥–µ–љ–љ–Њ—Б—В—М –њ–Њ–њ—Г–ї—П—Ж–Є–Є –љ–µ—Д—А–Њ–љ–Њ–≤ – –љ–Њ—А–Љ–∞–ї—М–љ—Л–µ (–Є–љ—В–∞–Ї—В–љ—Л–µ) –Ї–ї—Г–±–Њ—З–Ї–Є —З–µ—А–µ–і—Г—О—В—Б—П —Б –Є—И–µ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л–Љ–Є, –Ї–Њ–ї–ї–∞–±–Є—А–Њ–≤–∞–љ–љ—Л–Љ–Є, –∞—В—А–Њ—Д–Є—А–Њ–≤–∞–љ–љ—Л–Љ–Є –Є –≥–Є–њ–µ—А—В—А–Њ—Д–Є—А–Њ–≤–∞–љ–љ—Л–Љ–Є. –Т –њ–Њ—А–∞–ґ–µ–љ–љ—Л—Е –Ї–ї—Г–±–Њ—З–Ї–∞—Е –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–µ—В—Б—П —Г—В–Њ–ї—Й–µ–љ–Є–µ –Є —Б–Љ–Њ—А—Й–Є–≤–∞–љ–Є–µ —Б—В–µ–љ–Њ–Ї –Ї–∞–њ–Є–ї–ї—П—А–Њ–≤, —А–∞–Ј—А—Г—И–µ–љ–Є–µ –±–∞–Ј–∞–ї—М–љ–Њ–є –Љ–µ–Љ–±—А–∞–љ—Л. –У–Є–њ–µ—А—В—А–Њ—Д–Є—П –Є –≥–Є–њ–µ—А—Д—Г–љ–Ї—Ж–Є—П –Љ–µ–Ј–∞–љ–≥–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –њ—А–Є–≤–Њ–і—П—В –Ї –Є–Ј–±—Л—В–Њ—З–љ–Њ–Љ—Г –Њ—В–ї–Њ–ґ–µ–љ–Є—О –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ –≤ –Љ–µ–Ј–∞–љ–≥–Є–∞–ї—М–љ–Њ–Љ –Љ–∞—В—А–Є–Ї—Б–µ.
–Э–∞–Є–±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П –≤ –њ–Њ—З–µ—З–љ—Л—Е —Б–Њ—Б—Г–і–∞—Е. –°–ї–µ–і—Г–µ—В —З–µ—В–Ї–Њ —А–∞–Ј–і–µ–ї—П—В—М –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ–Њ—З–µ—З–љ—Л—Е –∞—А—В–µ—А–Є–є –Є –∞—А—В–µ—А–Є–Њ–ї. –°–њ–µ—Ж–Є—Д–Є—З–љ—Л–Љ —Б–Њ—Б—Г–і–Є—Б—В—Л–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –љ–µ—Д—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ —П–≤–ї—П–µ—В—Б—П –њ–Њ—А–∞–ґ–µ–љ–Є–µ —Б—А–µ–і–љ–Є—Е –Є –Љ–µ–ї–Ї–Є—Е –∞—А—В–µ—А–Є–є –≤–њ–ї–Њ—В—М –і–Њ –Љ–µ–ґ–і–Њ–ї—М–Ї–Њ–≤—Л—Е (–∞—А—В–µ—А–Є–Њ—Б–Ї–ї–µ—А–Њ–Ј). –Т –љ–Є—Е –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–µ—В—Б—П –≥–Є–њ–µ—А—В—А–Њ—Д–Є—П –Љ—Л—И–µ—З–љ–Њ–≥–Њ —Б–ї–Њ—П, —Г—В–Њ–ї—Й–µ–љ–Є–µ –Є —Д–Є–±—А–Њ–Ј –Є–љ—В–Є–Љ—Л –Ј–∞ —Б—З–µ—В –Њ—В–ї–Њ–ґ–µ–љ–Є–є –≥–Є–∞–ї–Є–љ–∞, —Г—В–Њ–ї—Й–µ–љ–Є–µ –Є —А–∞—Б—Й–µ–њ–ї–µ–љ–Є–µ –≤–љ—Г—В—А–µ–љ–љ–µ–є —Н–ї–∞—Б—В–Є—З–µ—Б–Ї–Њ–є –Љ–µ–Љ–±—А–∞–љ—Л. –Т —А–µ–Ј—Г–ї—М—В–∞—В–µ —Г—В–Њ–ї—Й–∞–µ—В—Б—П —Б—В–µ–љ–Ї–∞ —Б–Њ—Б—Г–і–∞ –Є —Г–Љ–µ–љ—М—И–∞–µ—В—Б—П –µ–≥–Њ –њ—А–Њ—Б–≤–µ—В [16,18,20].
–Р—А—В–µ—А–Є–Њ–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј–Њ–Љ –љ–∞–Ј—Л–≤–∞—О—В –Є–Ј–Љ–µ–љ–µ–љ–Є—П –∞—Д—Д–µ—А–µ–љ—В–љ—Л—Е –Є/–Є–ї–Є —Н—Д—Д–µ—А–µ–љ—В–љ—Л—Е –∞—А—В–µ—А–Є–Њ–ї, –Є–Љ–µ—О—Й–Є—Е (–≤ –Њ—В–ї–Є—З–Є–µ –Њ—В –∞—А—В–µ—А–Є–є) —В–Њ–ї—М–Ї–Њ 1–2 —Б–ї–Њ—П –≥–ї–∞–і–Ї–Њ–Љ—Л—И–µ—З–љ—Л—Е –Ї–ї–µ—В–Њ–Ї. –Р—А—В–µ—А–Є–Њ–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј —Е–∞—А–∞–Ї—В–µ—А–µ–љ –і–ї—П –Ј–ї–Њ–Ї–∞—З–µ—Б—В–≤–µ–љ–љ–Њ–є –Р–У, —Б–∞—Е–∞—А–љ–Њ–≥–Њ –і–Є–∞–±–µ—В–∞ –Є –љ–µ —П–≤–ї—П–µ—В—Б—П –Њ–±–ї–Є–≥–∞—В–љ—Л–Љ (—Е–Њ—В—П –љ–µ—А–µ–і–Ї–Њ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–µ—В—Б—П) –і–ї—П –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–Њ—З–µ–Ї [18].
–°—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П —В–∞–Ї–ґ–µ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П –≤ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–Є –њ–Њ—З–µ–Ї. –Э–∞–Є–±–Њ–ї–µ–µ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ —А–∞–Ј–≤–Є—В–Є—П —Д–Є–±—А–Њ–Ј–∞, —Б–Ї–Њ–њ–ї–µ–љ–Є–µ –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї, –њ–Њ—П–≤–ї–µ–љ–Є–µ –Ј–Њ–љ –Ї–∞–љ–∞–ї—М—Ж–µ–≤–Њ–є –∞—В—А–Њ—Д–Є–Є, —З–µ—А–µ–і—Г—О—Й–Є—Е—Б—П —Б –Ј–Њ–љ–∞–Љ–Є –≥–Є–њ–µ—А—В—А–Њ—Д–Є–Є –Ї–∞–љ–∞–ї—М—Ж–µ–≤. –Э–µ—А–µ–і–Ї–Њ –≤ –њ—А–Њ—Б–≤–µ—В–µ –Ї–∞–љ–∞–ї—М—Ж–µ–≤ –≤—Л—П–≤–ї—П—О—В—Б—П –±–µ–ї–Ї–Њ–≤—Л–µ —Ж–Є–ї–Є–љ–і—А—Л [16,18].
–Ь–µ—Е–∞–љ–Є–Ј–Љ—Л —А–∞–Ј–≤–Є—В–Є—П
–≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞
–Ь–µ—Е–∞–љ–Є–Ј–Љ—Л, –≤–Њ–≤–ї–µ—З–µ–љ–љ—Л–µ –≤ —А–∞–Ј–≤–Є—В–Є–µ –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞, —А–∞–Ј–і–µ–ї—П—О—В –љ–∞ –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–µ –Є –љ–µ–≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–µ, –Ї–Њ—В–Њ—А—Л–µ –≤–Ї–ї—О—З–∞—О—В –љ–µ–є—А–Њ–≥—Г–Љ–Њ—А–∞–ї—М–љ—Л–µ –Є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ —Д–∞–Ї—В–Њ—А—Л (—А–Є—Б. 5).
–У–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–є —Д–∞–Ї—В–Њ—А (–Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–∞—П –њ–µ—А–µ–≥—А—Г–Ј–Ї–∞ –Ы–Ц) —П–≤–ї—П–µ—В—Б—П –Њ–і–љ–Є–Љ –Є–Ј –љ–∞–Є–±–Њ–ї–µ–µ –Ј–љ–∞—З–Є–Љ—Л—Е –≤ —А–∞–Ј–≤–Є—В–Є–Є –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞. –Ю–і–љ–∞–Ї–Њ –њ–Њ––њ—А–µ–ґ–љ–µ–Љ—Г –Њ—Б—В–∞–µ—В—Б—П –і–Є—Б–Ї—Г—В–∞–±–µ–ї—М–љ—Л–Љ –≤–Њ–њ—А–Њ—Б, –Ї–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ –њ–µ—А–µ–≥—А—Г–Ј–Ї–∞ –Ы–Ц –њ—А–Є–≤–Њ–і–Є—В –Ї –µ–≥–Њ —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є—О. –ѓ–≤–ї—П–µ—В—Б—П –ї–Є –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Є–є —Б—В—А–µ—Б—Б –і–Њ—Б—В–∞—В–Њ—З–љ—Л–Љ —Г—Б–ї–Њ–≤–Є–µ–Љ –і–ї—П –≥–Є–њ–µ—А—В—А–Њ—Д–Є–Є –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–Њ–≤ –Є–ї–Є –µ–≥–Њ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ —П–≤–ї—П–µ—В—Б—П –љ–µ–њ—А—П–Љ—Л–Љ, –Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ—Л–Љ –љ–µ–є—А–Њ–≥—Г–Љ–Њ—А–∞–ї—М–љ—Л–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є? –†–µ–Ј—Г–ї—М—В–∞—В—Л —А—П–і–∞ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—В, —З—В–Њ –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Њ–µ —А–∞—Б—В—П–ґ–µ–љ–Є–µ –Ї–ї–µ—В–Њ–Ї –Љ–Є–Њ–Ї–∞—А–і–∞ –њ—А–Є–≤–Њ–і–Є—В –Ї —Г—Б–Є–ї–µ–љ–Є—О —Б–Є–љ—В–µ–Ј–∞ –±–µ–ї–Ї–∞ –Є —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є –љ–µ–Ї–Њ—В–Њ—А—Л—Е –≥–µ–љ–Њ–≤ —А–Њ—Б—В–∞ –±–µ–Ј —Г—З–∞—Б—В–Є—П –љ–µ—А–≤–љ—Л—Е –Є –≥—Г–Љ–Њ—А–∞–ї—М–љ—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤. –Ю–і–љ–Є–Љ –Є–Ј –≤–Њ–Ј–Љ–Њ–ґ–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ —В—А–∞–љ—Б–ї—П—Ж–Є–Є –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Њ–≥–Њ —Б—В–Є–Љ—Г–ї–∞ –≤ –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤ –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–µ —П–≤–ї—П–µ—В—Б—П –∞–Ї—В–Є–≤–∞—Ж–Є—П –њ—А–Њ—В–µ–Є–љ–Ї–Є–љ–∞–Ј–љ—Л—Е –њ—Г—В–µ–є. –†–Њ–ї—М –Љ–µ—Е–∞–љ–Њ—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –Љ–Њ–≥—Г—В —В–∞–Ї–ґ–µ –Є–≥—А–∞—В—М —В—А–∞–љ—Б–Љ–µ–Љ–±—А–∞–љ–љ—Л–µ –±–µ–ї–Ї–Є —Б–µ–Љ–µ–є—Б—В–≤–∞ –Є–љ—В–µ–≥—А–Є–љ–Њ–≤, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ—Л–µ –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –≤ –Љ–µ—Б—В–∞—Е –Ї–Њ–љ—В–∞–Ї—В–∞ –Љ–Є–Њ–Ї–∞—А–і–Є–Њ—Ж–Є—В–Њ–≤ —Б –≤–љ–µ–Ї–ї–µ—В–Њ—З–љ—Л–Љ –Љ–∞—В—А–Є–Ї—Б–Њ–Љ [36,37].
–Ь–µ–ґ–і—Г —В–µ–Љ –Љ–љ–Њ–≥–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–Є –љ–µ –њ—А–Є–Ј–љ–∞—О—В, —З—В–Њ –њ–µ—А–µ–≥—А—Г–Ј–Ї–∞ –Ы–Ц –Љ–Њ–ґ–µ—В –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ —Б—В–Є–Љ—Г–ї–Є—А–Њ–≤–∞—В—М —Б–Є–љ—В–µ–Ј –±–µ–ї–Ї–∞ –≤ –Љ–Є–Њ–Ї–∞—А–і–Є–Њ—Ж–Є—В–∞—Е –Є –≤—Л–Ј—Л–≤–∞—В—М –Ї–ї–µ—В–Њ—З–љ—Г—О –≥–Є–њ–µ—А—В—А–Њ—Д–Є—О. –Ю–љ–Є –њ–Њ–ї–∞–≥–∞—О—В, —З—В–Њ —Н—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ—А–Є –Њ–±—П–Ј–∞—В–µ–ї—М–љ–Њ–Љ —Г—З–∞—Б—В–Є–Є –љ–µ–≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є—Е –љ–µ–є—А–Њ–≥—Г–Љ–Њ—А–∞–ї—М–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤, –≤ –њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М –ї–Њ–Ї–∞–ї—М–љ–Њ–є (—В–Ї–∞–љ–µ–≤–Њ–є) —А–µ–љ–Є–љ––∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ––∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ–Њ–≤–Њ–є —Б–Є—Б—В–µ–Љ—Л (–†–Р–Р–°), —Н–љ–і–Њ—В–µ–ї–Є–љ–Њ–≤ –Є —Б–Є–Љ–њ–∞—В–Њ––∞–і—А–µ–љ–∞–ї–Њ–≤–Њ–є —Б–Є—Б—В–µ–Љ—Л (–°–Р–°) [7,38].
–Э–µ–≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–µ —Д–∞–Ї—В–Њ—А—Л —А–∞–Ј–≤–Є—В–Є—П –У–Ы–Ц –≤–Ї–ї—О—З–∞—О—В –љ–µ–є—А–Њ–≥—Г–Љ–Њ—А–∞–ї—М–љ—Л–µ –Є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ. –Э–∞–Є–±–Њ–ї—М—И–µ–µ –≤–љ–Є–Љ–∞–љ–Є–µ –≤ —А–∞–Ј–≤–Є—В–Є–Є –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞ —Г–і–µ–ї—П–µ—В—Б—П –†–Р–Р–°. –Р–Ї—В–Є–≤–∞—Ж–Є—П –ї–Њ–Ї–∞–ї—М–љ–Њ–є –†–Р–Р–° —Б–µ—А–і—Ж–∞ –≤—Л–Ј—Л–≤–∞–µ—В —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —Б–Њ–і–µ—А–ґ–∞–љ–Є—П –Р–ҐII, –Ї–Њ—В–Њ—А—Л–є, –≤–Њ–Ј–і–µ–є—Б—В–≤—Г—П –љ–∞ –Р–Ґ1–—А–µ—Ж–µ–њ—В–Њ—А—Л, –∞–Ї—В–Є–≤–Є—А—Г–µ—В –њ—А–Њ—В–µ–Є–љ–Ї–Є–љ–∞–Ј–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –Є –≤—Л–Ј—Л–≤–∞–µ—В –њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –≤ –Ї–∞—А–і–Є–Њ–Љ–Є–Њ—Ж–Є—В–∞—Е –Є –≥–ї–∞–і–Ї–Њ–Љ—Л—И–µ—З–љ—Л—Е –Ї–ї–µ—В–Ї–∞—Е –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е —Б–Њ—Б—Г–і–Њ–≤ [39,40]. –Р–ҐII —В–∞–Ї–ґ–µ —Г—З–∞—Б—В–≤—Г–µ—В –≤ —А–µ–≥—Г–ї—П—Ж–Є–Є –і–µ—П—В–µ–ї—М–љ–Њ—Б—В–Є —Д–Є–±—А–Њ–±–ї–∞—Б—В–Њ–≤ –≤ —Б–µ—А–і—Ж–µ: —Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В —Б–Є–љ—В–µ–Ј –±–µ–ї–Ї–Њ–≤ –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞ –Є –њ–Њ–і–∞–≤–ї—П–µ—В –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Љ–µ—В–∞–ї–ї–Њ–њ—А–Њ—В–µ–Є–љ–∞–Ј, –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л—Е –Ј–∞ –і–µ–≥—А–∞–і–∞—Ж–Є—О –±–µ–ї–Ї–Њ–≤ –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞ [40–42]. –Э–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Ј–∞–Љ–µ—В–Є—В—М, —З—В–Њ –њ–Њ–і–Њ–±–љ—Л–Љ —А–µ–≥—Г–ї–Є—А—Г—О—Й–Є–Љ –≤–ї–Є—П–љ–Є–µ–Љ –љ–∞ —Д–Є–±—А–Њ–±–ї–∞—Б—В—Л –Њ–±–ї–∞–і–∞–µ—В –Є –∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ. –Т —А–µ–Ј—Г–ї—М—В–∞—В–µ —Г–≤–µ–ї–Є—З–µ–љ–Є—П —Б–Є–љ—В–µ–Ј–∞ –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ—Л—Е –±–µ–ї–Ї–Њ–≤ –Є –љ–∞—А—Г—И–µ–љ–Є—П –µ–≥–Њ –і–µ–≥—А–∞–і–∞—Ж–Є–Є —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Д–Є–±—А–Њ–Ј – –Њ—В–ї–Є—З–Є—В–µ–ї—М–љ–∞—П —З–µ—А—В–∞ –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –≥–Є–њ–µ—А—В—А–Њ—Д–Є–Є [43,44].
–Ю–њ—А–µ–і–µ–ї–µ–љ–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –≤ —А–∞–Ј–≤–Є—В–Є–Є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–≤–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –≤ —Б–µ—А–і—Ж–µ –Є–Љ–µ–µ—В –°–Р–°. –≠–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л —Г–Ї–∞–Ј—Л–≤–∞—О—В –љ–∞ —В–Њ, —З—В–Њ –∞–Ї—В–Є–≤–∞—Ж–Є—П –°–Р–° —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –Ї–∞—А–і–Є–Њ—В—А–Њ—Д–Є—З–µ—Б–Ї–Є–Љ–Є —П–≤–ї–µ–љ–Є—П–Љ–Є –Є –Љ–Њ–ґ–µ—В —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞—В—М —А–∞–Ј–≤–Є—В–Є—О –У–Ы–Ц. –С–Њ–ї–µ–µ —В–Њ–≥–Њ, –њ–Њ –љ–µ–Ї–Њ—В–Њ—А—Л–Љ –і–∞–љ–љ—Л–Љ, –Ї–∞—В–µ—Е–Њ–ї–∞–Љ–Є–љ—Л –Љ–Њ–≥—Г—В –≤—Л–Ј—Л–≤–∞—В—М –љ–µ–Ї—А–Њ–Ј –Љ–Є–Њ—Ж–Є—В–Њ–≤, –∞ —В–∞–Ї–ґ–µ –Њ–Ї–∞–Ј—Л–≤–∞—В—М –≤–ї–Є—П–љ–Є–µ –љ–∞ –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ—Л–є –Љ–∞—В—А–Є–Ї—Б. –Ю–љ–Њ –Ј–∞–Ї–ї—О—З–∞–µ—В—Б—П, —Б –Њ–і–љ–Њ–є —Б—В–Њ—А–Њ–љ—Л, –≤ –Є–љ–і—Г–Ї—Ж–Є–Є —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Њ—Б—В–∞ —Д–Є–±—А–Њ–±–ї–∞—Б—В–Њ–≤ –Є —Г—Б–Є–ї–µ–љ–Є–Є —Д–Є–±—А–Њ–Ј–∞, —Б –і—А—Г–≥–Њ–є – –≤ —А–∞–Ј—А—Г—И–µ–љ–Є–Є –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤–Њ–є —Б–µ—В–Є –Є –њ–Њ—П–≤–ї–µ–љ–Є–Є —Г—З–∞—Б—В–Ї–Њ–≤ –љ–µ–Ї—А–Њ–Ј–∞. –Я—А–Є—З–Є–љ–љ–Њ–—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л–µ —Б–≤—П–Ј–Є –Љ–µ–ґ–і—Г –∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О –°–Р–° –Є —А–∞–Ј–≤–Є—В–Є–µ–Љ –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞ —В—А–µ–±—Г—О—В –і–∞–ї—М–љ–µ–є—И–µ–≥–Њ –Є–Ј—Г—З–µ–љ–Є—П. –Т–Њ–Ј–Љ–Њ–ґ–љ–Њ, —З—В–Њ –і–µ–є—Б—В–≤–Є–µ –Ї–∞—В–µ—Е–Њ–ї–∞–Љ–Є–љ–Њ–≤ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ –њ–Њ–Ј–Є—В–Є–≤–љ—Л–Љ —Е—А–Њ–љ–Њ—В—А–Њ–њ–љ—Л–Љ —Н—Д—Д–µ–Ї—В–Њ–Љ –Є–ї–Є —А–µ–∞–ї–Є–Ј—Г–µ—В—Б—П —З–µ—А–µ–Ј –∞–Ї—В–Є–≤–∞—Ж–Є—О –†–Р–Р–° [45–47].
–Ю–њ—А–µ–і–µ–ї–µ–љ–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –≤ —А–∞–Ј–≤–Є—В–Є–Є –Є–Ј–Љ–µ–љ–µ–љ–Є–є –Љ–Є–Њ–Ї–∞—А–і–∞ –њ—А–Є –Р–У –Љ–Њ–≥—Г—В –Є–Љ–µ—В—М —А—П–і –і—А—Г–≥–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤: —Б–Њ–Љ–∞—В–Њ—В—А–Њ–њ–љ—Л–є –≥–Њ—А–Љ–Њ–љ, –Є–љ—Б—Г–ї–Є–љ –Є –Є–љ—Б—Г–ї–Є–љ–Њ–њ–Њ–і–Њ–±–љ—Л–є —Д–∞–Ї—В–Њ—А, —Н–љ–і–Њ—В–µ–ї–Є–љ–1, –њ–∞—А–∞—В–≥–Њ—А–Љ–Њ–љ.
–Я—А–Њ–і–Њ–ї–ґ–∞–µ—В—Б—П –Є–Ј—Г—З–µ–љ–Є–µ —А–Њ–ї–Є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤ –≤ —А–∞–Ј–≤–Є—В–Є–Є –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞. –Я–Њ —А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ –љ–µ–Ї–Њ—В–Њ—А—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –Њ–љ–Є –Њ–њ—А–µ–і–µ–ї—П—О—В –і–Њ 30–60% –Ї–Њ–ї–µ–±–∞–љ–Є–є –Љ–∞—Б—Б—Л –Ы–Ц [48, 49]. –Э–∞–Є–±–Њ–ї—М—И–µ–µ —З–Є—Б–ї–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –Є –љ–∞–Є–±–Њ–ї–µ–µ –њ—А–Њ—В–Є–≤–Њ—А–µ—З–Є–≤—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л –њ–Њ–ї—Г—З–µ–љ—Л –њ—А–Є –Є–Ј—Г—З–µ–љ–Є–Є –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–∞ –≥–µ–љ–∞ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–њ—А–µ–≤—А–∞—Й–∞—О—Й–µ–≥–Њ —Д–µ—А–Љ–µ–љ—В–∞ (–Р–Я–§). –Т —А—П–і–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –±—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ I/D –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ —Б–≤—П–Ј–∞–љ —Б –У–Ы–Ц. –Ь–µ–ґ–і—Г —В–µ–Љ –і–∞–љ–љ—Л–µ, –њ–Њ–ї—Г—З–µ–љ–љ—Л–µ –≤ –њ–µ—А–≤—Л—Е —А–∞–±–Њ—В–∞—Е, –±—Л–ї–Є –≤ –њ–Њ—Б–ї–µ–і—Г—О—Й–µ–Љ –њ–Њ—Б—В–∞–≤–ї–µ–љ—Л –њ–Њ–і —Б–Њ–Љ–љ–µ–љ–Є–µ –њ–Њ —А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ –±–Њ–ї–µ–µ –Ї—А—Г–њ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є. –Ь–µ—В–∞––∞–љ–∞–ї–Є–Ј, –њ—А–Њ–≤–µ–і–µ–љ–љ—Л–є Staessen J.A. et al., –≤–Ї–ї—О—З–Є–≤—И–Є–є –≤ –Њ–±—Й–µ–є —Б–ї–Њ–ґ–љ–Њ—Б—В–Є 3285 –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –і–∞–ї –љ–µ—Г–±–µ–і–Є—В–µ–ї—М–љ—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л [50].
–Ш–Ј—Г—З–µ–љ–∞ –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –Љ–µ–ґ–і—Г –У–Ы–Ц –Є –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–Њ–≤ –≥–µ–љ–Њ–≤, –Ї–Њ–і–Є—А—Г—О—Й–Є—Е –і—А—Г–≥–Є–µ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В—Л –†–Р–Р–°. –Э–µ –±—Л–ї–Њ –њ–Њ–ї—Г—З–µ–љ–Њ —Г–±–µ–і–Є—В–µ–ї—М–љ—Л—Е –і–∞–љ–љ—Л—Е –≤ –њ–Њ–ї—М–Ј—Г –Ј–љ–∞—З–Є–Љ–Њ—Б—В–Є –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–∞ –≥–µ–љ–∞ —А–µ–љ–Є–љ–∞, –њ—А–Њ—В–Є–≤–Њ—А–µ—З–Є–≤—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л –і–∞–ї–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –Љ—Г—В–∞—Ж–Є–є –≥–µ–љ–∞ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≥–µ–љ–∞ (–Ь235–Ґ––њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ –Є –Ґ174–Ь––њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ). –Э–µ–њ–Њ–ї–љ–Њ—Б—В—М—О —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–∞ —А–Њ–ї—М –Р1166–°––њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–∞ –≥–µ–љ–∞ –Р–Ґ1–—А–µ—Ж–µ–њ—В–Њ—А–∞, –∞ –Ј–љ–∞—З–µ–љ–Є–µ G1675–Р––њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–∞ –Р–Ґ2–—А–µ—Ж–µ–њ—В–Њ—А–∞ –њ–Њ––њ—А–µ–ґ–љ–µ–Љ—Г —П–≤–ї—П–µ—В—Б—П –њ—А–µ–і–Љ–µ—В–Њ–Љ –∞–Ї—В–Є–≤–љ–Њ–≥–Њ –Є–Ј—Г—З–µ–љ–Є—П. –†—П–і –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–µ–є –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–ї–Є –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В—М –У–Ы–Ц –Є –і–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –Љ–Є–Њ–Ї–∞—А–і–∞ –Њ—В —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –∞–ї—М—В–µ—А–∞—Ж–Є–є –≥–µ–љ–∞ –∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ–—Б–Є–љ—В–∞–Ј—Л (–Њ–і–љ–∞–Ї–Њ –≤ –і—А—Г–≥–Є—Е —А–∞–±–Њ—В–∞—Е –њ–Њ–і–Њ–±–љ–Њ–є –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є –≤—Л—П–≤–ї–µ–љ–Њ –љ–µ –±—Л–ї–Њ).
–Т –њ–Њ—Б–ї–µ–і–љ–µ–µ –≤—А–µ–Љ—П –≤–µ–і—Г—В—Б—П –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ –Є–Ј—Г—З–µ–љ–Є—О —Б–≤—П–Ј–Є –У–Ы–Ц —Б –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–Њ–Љ –≥–µ–љ–Њ–≤, –Њ—В–≤–µ—З–∞—О—Й–Є—Е –Ј–∞ —Б–Є–љ—В–µ–Ј b2––∞–і—А–µ–љ–Њ—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤, –њ—А–Њ—В–µ–Є–љ–∞ G, –њ—А–Њ—В–µ–Є–љ–Ї–Є–љ–∞–Ј –Є —Н–љ–і–Њ—В–µ–ї–Є–љ–Њ–≤. –Я–Њ—П–≤–Є–ї–Є—Б—М –Є —А–µ–Ј—Г–ї—М—В–∞—В—Л –њ–µ—А–≤—Л—Е —А–∞–±–Њ—В –Њ —Б–≤—П–Ј–Є –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–Њ–≤ –≥–µ–љ–Њ–≤ NO–—Б–Є–љ—В–∞–Ј—Л (Glu298Asp, eNOS4b/4a) —Б –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –Њ—А–≥–∞–љ–Њ–≤––Љ–Є—И–µ–љ–µ–є –Р–У, —В—А–µ–±—Г—О—Й–Є–µ, –Њ–і–љ–∞–Ї–Њ, –і–∞–ї—М–љ–µ–є—И–µ–≥–Њ –Є–Ј—Г—З–µ–љ–Є—П [48].
–Ь–µ—Е–∞–љ–Є–Ј–Љ—Л —А–∞–Ј–≤–Є—В–Є—П
–≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–Њ—З–µ–Ї
–У–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–є —Д–∞–Ї—В–Њ—А. –Р–У —Г—Б–Ї–Њ—А—П–µ—В –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –•–Я–Э. –Я–Њ –і–∞–љ–љ—Л–Љ –Ї—А—Г–њ–љ—Л—Е –њ—А–Њ—Б–њ–µ–Ї—В–Є–≤–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, —Б—Г—Й–µ—Б—В–≤—Г–µ—В –і–Њ—Б—В–Њ–≤–µ—А–љ–∞—П –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –Љ–µ–ґ–і—Г —Г—А–Њ–≤–љ–µ–Љ –Р–Ф –Є —В–µ–Љ–њ–∞–Љ–Є –њ–∞–і–µ–љ–Є—П –°–Ъ–§ –љ–∞ —А–∞–љ–љ–Є—Е –Є –њ–Њ–Ј–і–љ–Є—Е —Б—В–∞–і–Є—П—Е –љ–µ—Д—А–Њ–њ–∞—В–Є–Є [51,52]. –Ъ–∞–Ї —Г–ґ–µ —Г–њ–Њ–Љ–Є–љ–∞–ї–Њ—Б—М –≤—Л—И–µ, –Љ–Њ–ґ–љ–Њ –≤—Л–і–µ–ї–Є—В—М –і–≤–∞ –њ—А–Є–љ—Ж–Є–њ–Є–∞–ї—М–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞, –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ –Ї–Њ—В–Њ—А–Њ–≥–Њ –Р–У —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —А–∞–Ј–≤–Є—В–Є—О –Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—О –•–Я–Э: 1) –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–∞—П –Є—И–µ–Љ–Є—П –Є 2) –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–∞—П –≥–Є–њ–µ—А—Д–Є–ї—М—В—А–∞—Ж–Є—П –Є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є—П [21]. –Ю–±–∞ –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞ –Є–Љ–µ—О—В –Ј–љ–∞—З–µ–љ–Є–µ –≤ –њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–Њ—З–µ–Ї: –њ—А–Є –њ–∞—В–Њ–Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –≤ –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ—З–Ї–µ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В –љ–µ—Д—А–Њ–љ—Л –Ї–∞–Ї —Б —П–≤–ї–µ–љ–Є—П–Љ–Є –Є—И–µ–Љ–Є–Є, —В–∞–Ї –Є —Б –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є –≥–Є–њ–µ—А–њ–µ—А—Д—Г–Ј–Є–Є (—А–Є—Б. 5).
–Я–µ—А–≤—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ («—В—А–∞–і–Є—Ж–Є–Њ–љ–љ—Л–є»), –≤–њ–µ—А–≤—Л–µ –њ—А–µ–і–ї–Њ–ґ–µ–љ–љ—Л–є Volhard –Є Fahr, –Ј–∞–Ї–ї—О—З–∞–µ—В—Б—П –≤ —А–∞–Ј–≤–Є—В–Є–Є —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –њ–Њ—З–µ—З–љ—Л—Е —Б–Њ—Б—Г–і–Њ–≤ (—Б–Ї–ї–µ—А–Њ–Ј) —Б —Б—Г–ґ–µ–љ–Є–µ–Љ –Є—Е –њ—А–Њ—Б–≤–µ—В–∞ –Є –њ–Њ—П–≤–ї–µ–љ–Є–µ–Љ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –Є—И–µ–Љ–Є–Є. –Т —А–µ–Ј—Г–ї—М—В–∞—В–µ —Н—В–Њ–≥–Њ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –°–Ъ–§ –Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –•–Я–Э [19].
–Ь–µ–ґ–і—Г —В–µ–Љ —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –і–∞–љ–љ—Л–µ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ —В–Њ–Љ, —З—В–Њ —А–∞–Ј–≤–Є—В–Є–µ –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –≤ –±–Њ–ї—М—И–µ–є —Б—В–µ–њ–µ–љ–Є —Б–≤—П–Ј–∞–љ–Њ —Б –њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ –≤–љ—Г—В—А–Є–Ї–ї—Г–±–Њ—З–Ї–Њ–≤—Л–Љ –і–∞–≤–ї–µ–љ–Є–µ–Љ, –љ–µ–ґ–µ–ї–Є —Б –Є—И–µ–Љ–Є–µ–є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤ [16,22]. –Э–∞ —А–∞–љ–љ–Є—Е —Н—В–∞–њ–∞—Е —А–∞–Ј–≤–Є—В–Є—П –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–Њ—З–µ–Ї –і–ї–Є—В–µ–ї—М–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ –Р–Ф –≤ —Б–Њ—З–µ—В–∞–љ–Є–Є —Б –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–Љ –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –∞—Г—В–Њ—А–µ–≥—Г–ї—П—Ж–Є–Є —В–Њ–љ—Г—Б–∞ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤—Л—Е –∞—А—В–µ—А–Є–Њ–ї –њ—А–Є–≤–Њ–і–Є—В –Ї —А–∞–Ј–≤–Є—В–Є—О —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –≥–Є–њ–µ—А–њ–µ—А—Д—Г–Ј–Є–Є –Є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є. –Т —Н—В–Є—Е —Г—Б–ї–Њ–≤–Є—П—Е —А–∞–Ј–≤–Є–≤–∞—О—В—Б—П —Б—В—А—Г–Ї—В—Г—А–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Ї–ї—Г–±–Њ—З–Ї–Њ–≤ (–≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј). –°—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ—А–µ—В–µ—А–њ–µ–≤–∞—О—В –Є –њ–Њ—З–µ—З–љ—Л–µ —Б–Њ—Б—Г–і—Л, –≤–Ї–ї—О—З–∞—П «–Є—И–µ–Љ–Є—З–µ—Б–Ї–Є–є» –Љ–µ—Е–∞–љ–Є–Ј–Љ —А–∞–Ј–≤–Є—В–Є—П –љ–µ—Д—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ [21,53,54].
–У–Є–±–µ–ї—М –Є–ї–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ –љ–∞—А—Г—И–µ–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–Є —З–∞—Б—В–Є –љ–µ—Д—А–Њ–љ–Њ–≤ –њ—А–Є–≤–Њ–і–Є—В –Ї —Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—О –Ї–Њ–Љ–њ–µ–љ—Б–∞—В–Њ—А–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ —Б —Ж–µ–ї—М—О –њ–Њ–і–і–µ—А–ґ–∞–љ–Є—П —Н–Ї—Б–Ї—А–µ—В–Њ—А–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї – –њ–Њ–≤—Л—И–µ–љ–Є—О —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –Р–Ф –Є –і–Є–ї–∞—В–∞—Ж–Є–Є –∞—Д—Д–µ—А–µ–љ—В–љ—Л—Е –∞—А—В–µ—А–Є–Њ–ї –Є–љ—В–∞–Ї—В–љ—Л—Е –љ–µ—Д—А–Њ–љ–Њ–≤. –Ю–і–љ–∞–Ї–Њ —Б —В–µ—З–µ–љ–Є–µ–Љ –≤—А–µ–Љ–µ–љ–Є –≥–Є–њ–µ—А—Д–Є–ї—М—В—А–∞—Ж–Є—П –њ—А–Є–≤–Њ–і–Є—В –Ї —Б—В—А—Г–Ї—В—Г—А–љ—Л–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ —Н—В–Є—Е –љ–µ—Д—А–Њ–љ–Њ–≤ –Є –і–∞–ї—М–љ–µ–є—И–µ–Љ—Г —Г—Б—Г–≥—Г–±–ї–µ–љ–Є—О –•–Я–Э – –∞–і–∞–њ—В–Є–≤–љ—Л–є –Њ—В–≤–µ—В —Б—В–∞–љ–Њ–≤–Є—В—Б—П –і–µ–Ј–∞–і–∞–њ—В–Є–≤–љ—Л–Љ, —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П «–њ–Њ—А–Њ—З–љ—Л–є –Ї—А—Г–≥» (—А–Є—Б. 3) [21,53,54].
–Э–µ–Љ–∞–ї–Њ–≤–∞–ґ–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –Є–Љ–µ–µ—В —А–µ–∞–Ї—Ж–Є—П –Љ–µ–Ј–∞–љ–≥–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –љ–∞ –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П. –Я–Њ–і –≤–ї–Є—П–љ–Є–µ–Љ –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Њ–є –љ–∞–≥—А—Г–Ј–Ї–Є –≤ –љ–Є—Е —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П —Б–Є–љ—В–µ–Ј –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ —В–Є–њ–Њ–≤ I –Є IV, —Д–Є–±—А–Њ–љ–µ–Ї—В–Є–љ–∞ –Є –ї–∞–Љ–Є–љ–Є–љ–∞. –≠—В–Њ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —Г–≤–µ–ї–Є—З–µ–љ–Є—О –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞ –Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—О –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ [55].
–Э–µ–≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–µ —Д–∞–Ї—В–Њ—А—Л. –Э–µ—Б–Љ–Њ—В—А—П –љ–∞ —В–Њ, —З—В–Њ –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П –Є–≥—А–∞—О—В —Б—Г—Й–µ—Б—В–≤–µ–љ–љ—Г—О —А–Њ–ї—М –≤ —А–∞–Ј–≤–Є—В–Є–Є –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–Њ—З–µ–Ї, —Г—А–Њ–≤–µ–љ—М –Р–Ф –ї–Є—И—М –љ–∞ 30% –Њ–њ—А–µ–і–µ–ї—П–µ—В —Б–Ї–Њ—А–Њ—Б—В—М —Б–љ–Є–ґ–µ–љ–Є—П –°–Ъ–§ [19].
–Ю–≥—А–Њ–Љ–љ–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –≤ —А–∞–Ј–≤–Є—В–Є–Є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є –≤ –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –њ—А–Є–і–∞–µ—В—Б—П –†–Р–Р–°. –Р–Ї—В–Є–≤–∞—Ж–Є—П –†–Р–Р–° –Њ–±–ї–∞–і–∞–µ—В —А—П–і–Њ–Љ –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є—Е –Є –љ–µ–≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є—Е —Н—Д—Д–µ–Ї—В–Њ–≤: 1) –њ–Њ–≤—Л—И–µ–љ–Є–µ —Б–Є—Б—В–µ–Љ–љ–Њ–≥–Њ –Р–Ф –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ –Љ–Њ—Й–љ–Њ–є –≤–∞–Ј–Њ–Ї–Њ–љ—Б—В—А–Є–Ї—Ж–Є–Є; 2) —А–∞–Ј–≤–Є—В–Є–µ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ –Ї–Њ–љ—Б—В—А–Є–Ї—Ж–Є–Є —Н—Д—Д–µ—А–µ–љ—В–љ–Њ–є –∞—А—В–µ—А–Є–Њ–ї—Л; 4) –љ–∞—А—Г—И–µ–љ–Є–µ –њ—А–Њ–љ–Є—Ж–∞–µ–Љ–Њ—Б—В–Є —Б—В–µ–љ–Ї–Є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤—Л—Е –Ї–∞–њ–Є–ї–ї—П—А–Њ–≤, –Ї–Њ–љ—В—А–∞–Ї—Ж–Є—П –Љ–µ–Ј–∞–љ–≥–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї, –≤–µ–і—Г—Й–∞—П –Ї —Г–Љ–µ–љ—М—И–µ–љ–Є—О –њ–ї–Њ—Й–∞–і–Є —Д–Є–ї—М—В—А–∞—Ж–Є–Њ–љ–љ–Њ–є –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–Є; 5) –∞–Ї—В–Є–≤–∞—Ж–Є—П —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Њ—Б—В–∞ –Є –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є–µ —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤; 6) –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є—П –Є–љ—В–µ—А—Б—В–Є—Ж–Є—П –Є–Љ–Љ—Г–љ–Њ–Ї–Њ–Љ–њ–µ—В–µ–љ—В–љ—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є; 7) —Г—Б–Є–ї–µ–љ–Є–µ –њ–µ—А–µ–Ї–Є—Б–љ–Њ–≥–Њ –Њ–Ї–Є—Б–ї–µ–љ–Є—П –ї–Є–њ–Є–і–Њ–≤ [56–58].
–Х—Й–µ —Б —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–Њ–≤ Bernard –°. –≤ 1859 –≥. –Є–Ј–≤–µ—Б—В–љ–Њ –Њ —Б—Г—Й–µ—Б—В–≤–Њ–≤–∞–љ–Є–Є —В–µ—Б–љ–Њ–є –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є –Љ–µ–ґ–і—Г –њ–Њ—З–µ—З–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–µ–є –Є –∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О –°–Р–°. –Э–∞ —Д–Њ–љ–µ —Г–≤–µ–ї–Є—З–µ–љ–љ–Њ–є —Б–Є–Љ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ—А–Њ–Є—Б—Е–Њ–і–Є—В: 1) –љ–∞—А—Г—И–µ–љ–Є–µ –њ–Њ—З–µ—З–љ–Њ–є –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є–Ї–Є –њ–Њ—Б—А–µ–і—Б—В–≤–Њ–Љ ?1––Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ–Њ–є –≤–∞–Ј–Њ–Ї–Њ–љ—Б—В—А–Є–Ї—Ж–Є–Є; 2) —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є—П —А–µ–љ–Є–љ–∞ –Є–Ј —О–Ї—Б—В–∞–≥–ї–Њ–Љ–µ—А—Г–ї—П—А–љ—Л—Е –Ї–ї–µ—В–Њ–Ї; 3) —Г—Б–Є–ї–µ–љ–Є–µ —А–µ–∞–±—Б–Њ—А–±—Ж–Є–Є –љ–∞—В—А–Є—П; 4) —Б—В–Є–Љ—Г–ї—П—Ж–Є—П –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П –Р–ҐII. –Ю–і–љ–∞–Ї–Њ –Ј–љ–∞—З–µ–љ–Є–µ –°–Р–° –≤ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–Є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є –њ–Њ–ї–љ–Њ—Б—В—М—О –љ–µ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ. –Я—А–µ–і–њ–Њ–ї–∞–≥–∞–µ—В—Б—П, —З—В–Њ –љ–∞ —А–∞–љ–љ–Є—Е —Н—В–∞–њ–∞—Е –≥–Є–њ–µ—А–∞–Ї—В–Є–≤–љ–Њ—Б—В—М –°–Р–° –≤—Л–Ј—Л–≤–∞–µ—В —А–∞–Ј–≤–Є—В–Є–µ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є, —В–Њ–≥–і–∞ –Ї–∞–Ї –љ–∞ –њ–Њ–Ј–і–љ–Є—Е —Б—В–∞–і–Є—П—Е, –љ–∞–њ—А–Њ—В–Є–≤, —Г–Љ–µ–љ—М—И–∞–µ—В –°–Ъ–§ –Є —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–є –њ–Њ—З–µ—З–љ—Л–є –њ–ї–∞–Ј–Љ–Њ—В–Њ–Ї [59–61].
–Т–∞–ґ–љ—Г—О —А–Њ–ї—М –≤ —А–∞–Ј–≤–Є—В–Є–Є –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є—Е –≤–љ—Г—В—А–Є–њ–Њ—З–µ—З–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є –Є–≥—А–∞—О—В –≤–∞–Ј–Њ–∞–Ї—В–Є–≤–љ—Л–µ —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л–µ —Д–∞–Ї—В–Њ—А—Л, —В–Њ—З–љ–µ–µ, –і–Є—Б–±–∞–ї–∞–љ—Б –Љ–µ–ґ–і—Г –≤–∞–Ј–Њ–і–Є–ї–∞—В–Є—А—Г—О—Й–Є–Љ–Є –Є –≤–∞–Ј–Њ–Ї–Њ–љ—Б—В—А–Є–Ї—В–Њ—А–љ—Л–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є. –Т —З–∞—Б—В–љ–Њ—Б—В–Є, –Є–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –њ–Њ–і–∞–≤–ї–µ–љ–Є–µ —Б–Є–љ—В–µ–Ј–∞ NO –њ—А–Є–≤–Њ–і–Є—В –Ї —А–∞–Ј–≤–Є—В–Є—О –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є, –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є –Є –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞.
–Я—А–Њ—В–µ–Є–љ—Г—А–Є—П —Б–∞–Љ–∞ –њ–Њ —Б–µ–±–µ, –њ–Њ––≤–Є–і–Є–Љ–Њ–Љ—Г, —П–≤–ї—П–µ—В—Б—П –Њ–і–љ–Є–Љ –Є–Ј —Д–∞–Ї—В–Њ—А–Њ–≤ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –љ–µ—Д—А–Њ–њ–∞—В–Є–Є. –Я—А–µ–і–њ–Њ–ї–∞–≥–∞–µ—В—Б—П, —З—В–Њ –±–µ–ї–Њ–Ї —Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є—О –Є–љ—В–µ—А—Б—В–Є—Ж–Є—П –Љ–Њ–љ–Њ—Ж–Є—В–∞–Љ–Є –Є –Ґ––ї–Є–Љ—Д–Њ—Ж–Є—В–∞–Љ–Є, –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —З–µ–≥–Њ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј. –Я–Њ–Љ–Є–Љ–Њ —Н—В–Њ–≥–Њ, –њ—А–Њ—Е–Њ–ґ–і–µ–љ–Є–µ –њ—А–Њ—В–µ–Є–љ–Њ–≤ —З–µ—А–µ–Ј –±–∞–Ј–∞–ї—М–љ—Г—О –Љ–µ–Љ–±—А–∞–љ—Г –њ—А–Є–≤–Њ–і–Є—В –Ї –µ–µ –і–∞–ї—М–љ–µ–є—И–µ–Љ—Г –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О [62,63].
–Я–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –љ–∞—А—Г—И–µ–љ–Є—П –Њ–±–Љ–µ–љ–∞ –ї–Є–њ–Є–і–Њ–≤ (–≥–Є–њ–µ—А– –Є –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є—П) —В–∞–Ї–ґ–µ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В —А–∞–Ј–≤–Є—В–Є—О –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–Њ—З–µ–Ї. –°–Њ–≥–ї–∞—Б–љ–Њ –Њ–і–љ–Њ–Љ—Г –Є–Ј –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–Є–є –ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–і—Л –љ–Є–Ј–Ї–Њ–є –њ–ї–Њ—В–љ–Њ—Б—В–Є (–Ы–Я–Э–Я), –Ј–∞—Е–≤–∞—З–µ–љ–љ—Л–µ –Љ–µ–Ј–∞–љ–≥–Є–∞–ї—М–љ—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є, —Б—В–Є–Љ—Г–ї–Є—А—Г—О—В –Є—Е –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є—О –Є –≤—Л—А–∞–±–Њ—В–Ї—Г —Е–µ–Љ–Њ—В–∞–Ї—Б–Є—З–µ—Б–Ї–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤. –Ю–Ї–Є—Б–ї–µ–љ–Є–µ –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–Љ–Є –Є –Љ–µ–Ј–∞–љ–≥–Є–∞–ї—М–љ—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є –Ы–Я–Э–Я –њ—А–Є–≤–Њ–і–Є—В –Ї –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—О —В—Г—З–љ—Л—Е –Ї–ї–µ—В–Њ–Ї, –Ї–Њ—В–Њ—А—Л–µ –≤—Л—Б–≤–Њ–±–Њ–ґ–і–∞—О—В —Ж–Є—В–Њ–Ї–Є–љ—Л, —Д–∞–Ї—В–Њ—А—Л —А–Њ—Б—В–∞ –Є –і—А—Г–≥–Є–µ –≤–µ—Й–µ—Б—В–≤–∞, –≤ —Ж–µ–ї–Њ–Љ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—Й–Є–µ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—О –Є —Н–Ї—Б–њ–∞–љ—Б–Є–Є –Љ–µ–Ј–∞–љ–≥–Є—П [64].
–†–µ–Ј—Г–ї—М—В–∞—В—Л –і–Њ—Б—В–∞—В–Њ—З–љ–Њ –±–Њ–ї—М—И–Њ–≥–Њ —З–Є—Б–ї–∞ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ–Ј–≤–Њ–ї—П—О—В –њ—А–µ–і–њ–Њ–ї–∞–≥–∞—В—М —Г—З–∞—Б—В–Є–µ –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤ –≤ —А–∞–Ј–≤–Є—В–Є–Є –Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–Є –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–Њ—З–µ–Ї. –Я–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤–∞—П –≥–Є–њ–µ—А—Д–Є–ї—М—В—А–∞—Ж–Є—П, –њ–Њ–≤—Л—И–µ–љ–љ–Њ–µ –њ–Њ—З–µ—З–љ–Њ–µ —Б–Њ—Б—Г–і–Є—Б—В–Њ–µ —Б–Њ–њ—А–Њ—В–Є–≤–ї–µ–љ–Є–µ –Є –Ь–Є–Р –љ–µ—А–µ–і–Ї–Њ –≤—Л—П–≤–ї—П—О—В—Б—П –і–∞–ґ–µ —Г –Ј–і–Њ—А–Њ–≤—Л—Е –ї–Є—Ж, —А–Њ–і–Є—В–µ–ї–Є –Ї–Њ—В–Њ—А—Л—Е —Б—В—А–∞–і–∞—О—В –Р–У [65,66]. –Ш–Ј—Г—З–µ–љ –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ —Ж–µ–ї–Њ–≥–Њ —А—П–і–∞ –≥–µ–љ–Њ–≤ –љ–∞ –њ—А–µ–і–Љ–µ—В –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є —Б —А–∞–Ј–≤–Є—В–Є–µ–Љ –љ–µ—Д—А–Њ–њ–∞—В–Є–Є. –Э–∞–Є–±–Њ–ї—М—И–Є–є –Є–љ—В–µ—А–µ—Б –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–µ–є –≤—Л–Ј—Л–≤–∞–µ—В –Є–Ј—Г—З–µ–љ–Є–µ –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–∞ –≥–µ–љ–Њ–≤ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–Њ–≤ –†–Р–Р–°, –≤ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є I/D –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ –≥–µ–љ–∞ –Р–Я–§.
–Т —А—П–і–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є (–Ј–∞ –љ–µ–Ї–Њ—В–Њ—А—Л–Љ –Є—Б–Ї–ї—О—З–µ–љ–Є–µ–Љ) –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –≥–µ–љ–Њ—В–Є–њ DD –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–∞–љ —Б –±–Њ–ї–µ–µ –±—Л—Б—В—А—Л–Љ–Є —В–µ–Љ–њ–∞–Љ–Є —Б–љ–Є–ґ–µ–љ–Є—П –њ–Њ—З–µ—З–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –Є –љ–µ–і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–µ–є [67–69]. –Т –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е —А–∞–±–Њ—В–∞—Е –±—Л–ї–∞ –њ–Њ–Ї–∞–Ј–∞–љ–∞ –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –Љ–µ–ґ–і—Г –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–Њ–Љ –Ь235–Ґ –≥–µ–љ–∞ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–∞ –Є —А–∞–Ј–≤–Є—В–Є–µ–Љ –љ–µ—Д—А–Њ–њ–∞—В–Є–Є [70]. Coll –Х. et al. –љ–µ–і–∞–≤–љ–Њ –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ A1166C –≥–µ–љ–∞ –Р–Ґ1–—А–µ—Ж–µ–њ—В–Њ—А–∞ —Б–≤—П–Ј–∞–љ —Б –њ—А–µ–і—А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ–Њ—Б—В—М—О –Ї –±–Њ–ї–µ–µ –±—Л—Б—В—А–Њ–Љ—Г –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—О —В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є –•–Я–Э —А–∞–Ј–ї–Є—З–љ–Њ–є —Н—В–Є–Њ–ї–Њ–≥–Є–Є [71]. –Ю–і–љ–∞–Ї–Њ –≤ –і—А—Г–≥–Є—Е —А–∞–±–Њ—В–∞—Е –њ–Њ–і–Њ–±–љ–Њ–є –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ –љ–µ –±—Л–ї–Њ. –Я—А–Њ–і–Њ–ї–ґ–∞–µ—В—Б—П –Є–Ј—Г—З–µ–љ–Є–µ –Ј–љ–∞—З–µ–љ–Є—П –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ–∞ –≥–µ–љ–Њ–≤ –∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ–—Б–Є–љ—В–∞–Ј—Л, ?3–—Б—Г–±—К–µ–і–Є–љ–Є—Ж—Л –њ—А–Њ—В–µ–Є–љ–∞ G –Є –Њ–Ї—Б–Є–і–∞ –∞–Ј–Њ—В–∞ (NO).
–Я–∞—А–∞–ї–ї–µ–ї–Є–Ј–Љ —А–∞–Ј–≤–Є—В–Є—П –Є —В–Њ—З–Ї–Є —Б–Њ–њ—А–Є–Ї–Њ—Б–љ–Њ–≤–µ–љ–Є—П
—Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Є –њ–Њ—З–µ—З–љ–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞
–°–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–є –Є –њ–Њ—З–µ—З–љ—Л–є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ –љ–∞—Е–Њ–і—П—В—Б—П –≤ —В–µ—Б–љ–Њ–є –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є (—А–Є—Б. 6). –Э–µ—Д—А–Њ–∞–љ–≥–Є–Њ—Б–Ї–ї–µ—А–Њ–Ј –Є —А–∞–Ј–≤–Є—В–Є–µ –•–Я–Э –љ–µ—А–µ–і–Ї–Њ –љ–∞–±–ї—О–і–∞–µ—В—Б—П —Г –±–Њ–ї—М–љ—Л—Е –°–°–Ч. –Т —Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –°–°–Ч —П–≤–ї—П—О—В—Б—П –Њ—Б–љ–Њ–≤–љ–Њ–є –њ—А–Є—З–Є–љ–Њ–є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б —В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є –•–Я–Э, –∞ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–∞—П —Б–Љ–µ—А—В–љ–Њ—Б—В—М —Б—А–µ–і–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–∞ –і–Є–∞–ї–Є–Ј–µ –≤ –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ —А–∞–Ј –≤—Л—И–µ, —З–µ–Љ –≤ –Њ–±—Й–µ–є –њ–Њ–њ—Г–ї—П—Ж–Є–Є. –Ю–і–љ–∞–Ї–Њ —Б–≤—П–Ј—М –Љ–µ–ґ–і—Г –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є —Б–µ—А–і—Ж–∞ –Є –њ–Њ—З–µ–Ї –њ—А–Њ—Б–ї–µ–ґ–Є–≤–∞–µ—В—Б—П –љ–∞ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –±–Њ–ї–µ–µ —А–∞–љ–љ–Є—Е —Н—В–∞–њ–∞—Е. –Ю —В–µ—Б–љ–Њ–є —Б–≤—П–Ј–Є –Љ–µ–ґ–і—Г —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–Љ –Є –њ–Њ—З–µ—З–љ—Л–Љ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–Њ–Љ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В —Б—Г—Й–µ—Б—В–≤–Њ–≤–∞–љ–Є–µ —Ж–µ–ї–Њ–≥–Њ —А—П–і–∞ –Ї–ї–Є–љ–Є–Ї–Њ––ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л—Е –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є, —П–≤–ї—П—О—Й–Є—Е—Б—П –µ–і–Є–љ—Л–Љ–Є –Љ–∞—А–Ї–µ—А–∞–Љ–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞ –Є –њ–Њ—З–µ–Ї.
–У–Є–њ–µ—А—В—А–Њ—Д–Є—П –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ —П–≤–ї—П–µ—В—Б—П –і–Њ–Ї–∞–Ј–∞–љ–љ—Л–Љ –њ—А–Њ–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є –Ј–∞–±–Њ–ї–µ–≤–∞–µ–Љ–Њ—Б—В–Є –Є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є. –Т–Њ –§—А–∞–Љ–Є–љ–≥–µ–Љ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М –У–Ы–Ц –±—Л–ї–∞ –≤ 3–4 —А–∞–Ј–∞ –≤—Л—И–µ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –•–Я–Э, —З–µ–Љ —Б –љ–Њ—А–Љ–∞–ї—М–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–µ–є –њ–Њ—З–µ–Ї –њ—А–Є —Б–Њ–њ–Њ—Б—В–∞–≤–Є–Љ–Њ–Љ —Г—А–Њ–≤–љ–µ –Р–Ф [25]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –У–Ы–Ц —П–≤–ї—П–µ—В—Б—П –њ—А–µ–і–Є–Ї—В–Њ—А–Њ–Љ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л—Е —Б–Њ–±—Л—В–Є–є —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –•–Я–Э. –С–Њ–ї–µ–µ —З–µ–Љ 2/3 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –У–Ы–Ц, –љ–∞—Е–Њ–і—П—Й–Є—Е—Б—П –љ–∞ –і–Є–∞–ї–Є–Ј–µ, —Г–Љ–Є—А–∞—О—В –Њ—В —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –Є–ї–Є –≤–љ–µ–Ј–∞–њ–љ–Њ–є —Б–Љ–µ—А—В–Є, –∞ 5––ї–µ—В–љ—П—П —Б–Љ–µ—А—В–љ–Њ—Б—В—М –≤–Њ–Ј—А–∞—Б—В–∞–µ—В —Б 23 –і–Њ 52%, –µ—Б–ї–Є –Є–љ–і–µ–Ї—Б –Љ–∞—Б—Б—Л –Љ–Є–Њ–Ї–∞—А–і–∞ –Ы–Ц –њ—А–µ–≤—Л—И–∞–µ—В 125 –≥/–Љ2 [72].
–Я—А–Њ—В–µ–Є–љ—Г—А–Є—П —П–≤–ї—П–µ—В—Б—П —Б–∞–Љ—Л–Љ —Б–Є–ї—М–љ—Л–Љ –њ—А–µ–і–Є–Ї—В–Њ—А–Њ–Љ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –Є –љ–µ–і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є –Є —А–∞–Ј–≤–Є—В–Є—П —В–µ—А–Љ–Є–љ–∞–ї—М–љ–Њ–є –•–Я–Э. –£ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Є—Б—Е–Њ–і–љ–Њ–є –њ—А–Њ—В–µ–Є–љ—Г—А–Є–µ–є <1,9 –≥/—Б—Г—В. —В–µ–Љ–њ—Л —Б–љ–Є–ґ–µ–љ–Є—П –°–Ъ–§ –љ–µ –њ—А–µ–≤—Л—Б–Є–ї–Є 5% –≤ –≥–Њ–і. –£ –±–Њ–ї—М–љ—Л—Е —Б —Г—А–Њ–≤–љ–µ–Љ –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є >3,9 –≥/—Б—Г—В. –Њ—В–Љ–µ—З–µ–љ–Њ —Б–љ–Є–ґ–µ–љ–Є–µ –°–Ъ–§ –±–Њ–ї–µ–µ —З–µ–Љ –љ–∞ 10 –Љ–ї/–Љ–Є–љ./1,73 –Љ2 —Б —А–Є—Б–Ї–Њ–Љ —А–∞–Ј–≤–Є—В–Є—П –•–Я–Э >30% –Ј–∞ 3 –≥–Њ–і–∞ [73,74]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –њ—А–Њ—В–µ–Є–љ—Г—А–Є—П —П–≤–ї—П–µ—В—Б—П –Љ–∞—А–Ї–µ—А–Њ–Љ –њ–Њ–≤—Л—И–µ–љ–љ–Њ–≥–Њ —А–Є—Б–Ї–∞ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є –Ј–∞–±–Њ–ї–µ–≤–∞–µ–Љ–Њ—Б—В–Є –Є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є. –Т–њ–µ—А–≤—Л–µ —Н—В–Њ—В —Д–∞–Ї—В –±—Л–ї —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ –≤–Њ –§—А–∞–Љ–Є–љ–≥–µ–Љ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –Є –≤–њ–Њ—Б–ї–µ–і—Б—В–≤–Є–Є –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ –≤ —А—П–і–µ –і—А—Г–≥–Є—Е —А–∞–±–Њ—В [75,76].
–Ф–∞–љ–љ—Л–µ, –њ–Њ–ї—Г—З–µ–љ–љ—Л–µ –≤ —Е–Њ–і–µ –Ї—А—Г–њ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—В, —З—В–Њ –њ–Њ–≤—Л—И–µ–љ–љ—Л–є —Г—А–Њ–≤–µ–љ—М –Ї—А–µ–∞—В–Є–љ–Є–љ–∞ –Ї—А–Њ–≤–Є —П–≤–ї—П–µ—В—Б—П –Љ–Њ—Й–љ—Л–Љ –љ–µ–Ј–∞–≤–Є—Б–Є–Љ—Л–Љ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ —А–Є—Б–Ї–∞. –Ґ–∞–Ї, –≤ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є HOT —Г—А–Њ–≤–µ–љ—М –Ї—А–µ–∞—В–Є–љ–Є–љ–∞ –Њ–Ї–∞–Ј–∞–ї—Б—П –љ–∞–Є–±–Њ–ї–µ–µ –Ј–љ–∞—З–Є–Љ—Л–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ —А–Є—Б–Ї–∞ —Б–Љ–µ—А—В–љ–Њ—Б—В–Є –≤ —Б—А–∞–≤–љ–µ–љ–Є–Є —Б –і—А—Г–≥–Є–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є [77]. –С–Њ–ї—М—И–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –≤ –Ї–∞—З–µ—Б—В–≤–µ –њ—А–µ–і–Є–Ї—В–Њ—А–∞ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ —А–Є—Б–Ї–∞ –Є–Љ–µ–µ—В —В–∞–Ї–ґ–µ –Ї–ї–Є—А–µ–љ—Б –Ї—А–µ–∞—В–Є–љ–Є–љ–∞, –Є–ї–Є –°–Ъ–§.
–Ь–Є–Ї—А–Њ–∞–ї—М–±—Г–Љ–Є–љ—Г—А–Є—П, —Б—Г–±–Ї–ї–Є–љ–Є—З–µ—Б–Ї–∞—П —Н–Ї—Б–Ї—А–µ—Ж–Є—П –∞–ї—М–±—Г–Љ–Є–љ–∞, –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–∞–љ–∞ —Б –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ–Љ –љ–µ—Д—А–Њ–њ–∞—В–Є–Є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –Є –љ–µ–і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є —Н—В–Є–Њ–ї–Њ–≥–Є–Є. –Ь–Є–Р —А–∞—Б—Ж–µ–љ–Є–≤–∞–µ—В—Б—П –Ї–∞–Ї –Љ–∞—А–Ї–µ—А —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є –њ–Њ—З–µ—З–љ–Њ–є –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є–Ї–Є, –∞ —В–∞–Ї–ґ–µ —А–∞–љ–љ–Є—Е —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є, –Ї–∞–Ї –њ—А–µ–і–Є–Ї—В–Њ—А —А–∞–Ј–≤–Є—В–Є—П –•–Я–Э [65,78–81].
–Т –њ–Њ—Б–ї–µ–і–љ–Є–µ –≥–Њ–і—Л –Ї –Ь–Є–Р –Њ–±—А–∞—Й–µ–љ–Њ –Њ—Б–Њ–±–Њ–µ –≤–љ–Є–Љ–∞–љ–Є–µ. –Ю–љ–Њ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ –≤—Л—П–≤–ї–µ–љ–љ–Њ–є –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М—О –Љ–µ–ґ–і—Г –њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ —Г—А–Њ–≤–љ–µ–Љ —Н–Ї—Б–Ї—А–µ—Ж–Є–Є –∞–ї—М–±—Г–Љ–Є–љ–∞ –Є —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є —А–Є—Б–Ї–∞, –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є—П–Љ–Є –Є —Б–Љ–µ—А—В–љ–Њ—Б—В—М—О [81].
–Ь–Є–Р —П–≤–ї—П–µ—В—Б—П –Њ—В—А–∞–ґ–µ–љ–Є–µ–Љ —А–∞–љ–љ–Є—Е —Б—В–∞–і–Є–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є. –С–Њ–ї–µ–µ —В–Њ–≥–Њ, –Є–љ–Њ–≥–і–∞ –Ь–Є–Р –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–µ—В—Б—П —Г –Ј–і–Њ—А–Њ–≤—Л—Е –ї–Є—Ж –Є –њ—А–µ–і—И–µ—Б—В–≤—Г–µ—В —А–∞–Ј–≤–Є—В–Є—О –Р–У. –У–Є—Б—В–Њ–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –љ–µ –≤—Л—П–≤–ї—П—О—В —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є —Д–Є–ї—М—В—А–∞—Ж–Є–Њ–љ–љ–Њ–≥–Њ –±–∞—А—М–µ—А–∞ –Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є—П —Г –±–Њ–ї—М–љ—Л—Е –Р–У —Б –Ь–Є–Р [82]. –Ю–і–љ–∞–Ї–Њ –њ—А–Є—Б—Г—В—Б—В–≤–Є–µ –Ь–Є–Р –Ї–Њ—А—А–µ–ї–Є—А—Г–µ—В —Б –љ–∞–ї–Є—З–Є–µ–Љ –≥–Є–њ–µ—А—Д–Є–ї—М—В—А–∞—Ж–Є–Є –Є –≥–Є–њ–µ—А–њ–µ—А—Д—Г–Ј–Є–Є. –Ь–Є–Р –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–∞–љ–∞ —Б —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є —А–Є—Б–Ї–∞ (–Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В—М—О, –≥–Є–њ–µ—А–ї–Є–њ–Є–і–µ–Љ–Є–µ–є, –Є–Ј–±—Л—В–Њ—З–љ–Њ–є –Љ–∞—Б—Б–Њ–є —В–µ–ї–∞) —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Р–У –Є –і–∞–ґ–µ –Ј–і–Њ—А–Њ–≤—Л—Е –ї–Є—Ж. –Ь–Є–Р –Ї–Њ—А—А–µ–ї–Є—А—Г–µ—В —Б —Г—А–Њ–≤–љ–µ–Љ –Р–Ф –Є –Њ–±—А–∞—В–љ–Њ –Ї–Њ—А—А–µ–ї–Є—А—Г–µ—В —Б–Њ —Б—В–µ–њ–µ–љ—М—О –љ–Њ—З–љ–Њ–≥–Њ —Б–љ–Є–ґ–µ–љ–Є—П –Р–Ф. –£ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Ь–Є–Р —З–∞—Й–µ –≤—Л—П–≤–ї—П—О—В –У–Ы–Ц, –Ї–Њ–љ—Ж–µ–љ—В—А–Є—З–µ—Б–Ї—Г—О –≥–µ–Њ–Љ–µ—В—А–Є—О –Ы–Ц. –С–Њ–ї–µ–µ —В–Њ–≥–Њ, –љ–∞–ї–Є—З–Є–µ –Ь–Є–Р, –≥–Є–њ–µ—А–њ–µ—А—Д—Г–Ј–Є–Є –Є –≥–Є–њ–µ—А—Д–Є–ї—М—В—А–∞—Ж–Є–Є –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–∞–љ–Њ —Б —А–∞–љ–љ–Є–Љ–Є –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є —Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є –Є –і–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –Ы–Ц [83]. –Э–∞–Љ–Є –±—Л–ї–∞ –Њ–±–љ–∞—А—Г–ґ–µ–љ–∞ –љ–µ–њ—А–µ—А—Л–≤–љ–∞—П –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –Љ–µ–ґ–і—Г –Љ–∞—Б—Б–Њ–є –Љ–Є–Њ–Ї–∞—А–і–∞ –Ы–Ц –Є —Г—А–Њ–≤–љ–µ–Љ —Н–Ї—Б–Ї—А–µ—Ж–Є–Є –∞–ї—М–±—Г–Љ–Є–љ–∞ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –±–µ–Ј –У–Ы–Ц –Є –Ь–Є–Р —Б –љ–µ–і–∞–≤–љ–Њ –≤—Л—П–≤–ї–µ–љ–љ–Њ–є –Р–У [23]. –Я–Њ–Ї–∞–Ј–∞–љ–Њ —В–∞–Ї–ґ–µ –љ–∞–ї–Є—З–Є–µ –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є –Љ–µ–ґ–і—Г –Ь–Є–Р –Є –°–°–Ч, —В–∞–Ї–Є–Љ–Є –Ї–∞–Ї –Ш–С–°, –Ш–Ь, —Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Є –•–°–Э, –Њ–і–љ–∞–Ї–Њ –Њ–љ–∞ –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ —Б–ї–∞–±–∞ [84].
–°–Њ–≥–ї–∞—Б–љ–Њ –Њ–і–љ–Њ–є –Є–Ј –≥–Є–њ–Њ—В–µ–Ј –Ь–Є–Р —П–≤–ї—П–µ—В—Б—П –њ–Њ—З–µ—З–љ—Л–Љ –≤—Л—А–∞–ґ–µ–љ–Є–µ–Љ –≥–µ–љ–µ—А–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–є —Б–Њ—Б—Г–і–Є—Б—В–Њ–є, –Є–ї–Є —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є. –Т –і–µ–є—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є, —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П —П–≤–ї—П–µ—В—Б—П –Њ–±—Й–Є–Љ –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–Љ —А–∞–Ј–≤–Є—В–Є—П –°–°–Ч –Є –Њ–і–љ–Є–Љ –Є–Ј —А–∞–љ–љ–Є—Е —Н—В–∞–њ–Њ–≤ –∞—В–µ—А–Њ–≥–µ–љ–µ–Ј–∞ [85]. –ѓ–≤–ї—П—П—Б—М —Г–љ–Є–≤–µ—А—Б–∞–ї—М–љ—Л–Љ –њ—А–Њ—Ж–µ—Б—Б–Њ–Љ –≤ —Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–Є—Б—В–µ–Љ–µ, –Ј–∞—В—А–∞–≥–Є–≤–∞—П, –≤ —В–Њ–Љ —З–Є—Б–ї–µ, –Ї–∞–њ–Є–ї–ї—П—А—Л –Ї–ї—Г–±–Њ—З–Ї–Њ–≤, —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Љ–Њ–ґ–µ—В –Њ–±—К—П—Б–љ–Є—В—М –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –Љ–µ–ґ–і—Г –Ь–Є–Р, —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є —А–Є—Б–Ї–∞ –Є —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є –Ј–∞–±–Њ–ї–µ–≤–∞–µ–Љ–Њ—Б—В—М—О.
–Т–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –Љ–µ–ґ–і—Г —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–Љ –Є –њ–Њ—З–µ—З–љ—Л–Љ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–Њ–Љ —Б–Њ—Е—А–∞–љ—П–µ—В—Б—П –Є –љ–∞ –±–Њ–ї–µ–µ –њ–Њ–Ј–і–љ–Є—Е —Н—В–∞–њ–∞—Е.
–Э–∞—А—Г—И–µ–љ–Є—П –њ–Њ—З–µ—З–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –Є–≥—А–∞—О—В –≤–∞–ґ–љ—Г—О —А–Њ–ї—М –≤ –њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –•–°–Э [25]. –°—Г—Й–µ—Б—В–≤—Г–µ—В –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–Є–µ –Њ —В–Њ–Љ, —З—В–Њ —Б–Њ—Е—А–∞–љ–љ–Њ—Б—В—М —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї —П–≤–ї—П–µ—В—Б—П –љ–∞–Є–±–Њ–ї–µ–µ –≤–∞–ґ–љ—Л–Љ —Д–∞–Ї—В–Њ—А–Њ–Љ, –Њ–њ—А–µ–і–µ–ї—П—О—Й–Є–Љ –Ї–Њ–Љ–њ–µ–љ—Б–∞—Ж–Є—О –±–Њ–ї—М–љ—Л—Е —Б –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О –Ї—А–Њ–≤–Њ–Њ–±—А–∞—Й–µ–љ–Є—П [86]. –£—Е—Г–і—И–µ–љ–Є–µ –Ј–∞–њ–Њ–ї–љ–µ–љ–Є—П –∞—А—В–µ—А–Є–є –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є –Њ–і–Є–љ –Є–Ј –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤, —Б—В–Є–Љ—Г–ї–Є—А—Г—О—Й–Є—Е –Ј–∞–і–µ—А–ґ–Ї—Г –њ–Њ—З–Ї–∞–Љ–Є –љ–∞—В—А–Є—П –Є –ґ–Є–і–Ї–Њ—Б—В–Є. –Ю–і–љ–Њ–≤—А–µ–Љ–µ–љ–љ–Њ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –љ–µ–є—А–Њ–≥—Г–Љ–Њ—А–∞–ї—М–љ–∞—П –∞–Ї—В–Є–≤–∞—Ж–Є—П —Б —Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –†–Р–Р–° –Є –°–Р–°, –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–Є–µ–Љ –≤–∞–Ј–Њ–њ—А–µ—Б—Б–Є–љ–∞ –Є —Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ –њ—А–Њ–і—Г–Ї—Ж–Є–Є —Н–љ–і–Њ—В–µ–ї–Є–љ–∞ [87]. –£–ґ–µ –љ–∞ —А–∞–љ–љ–Є—Е —Б—В–∞–і–Є—П—Е –•–°–Э –Њ—В–Љ–µ—З–∞—О—В—Б—П –љ–∞—А—Г—И–µ–љ–Є—П —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї —Б–Њ —Б–љ–Є–ґ–µ–љ–Є–µ–Љ —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–≥–Њ –њ–Њ—З–µ—З–љ–Њ–≥–Њ —А–µ–Ј–µ—А–≤–∞ [88,89]. –Я–Њ –Љ–µ—А–µ —Н–≤–Њ–ї—О—Ж–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–µ —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –њ–Њ—З–µ—З–љ–Њ–≥–Њ –њ–ї–∞–Ј–Љ–Њ—В–Њ–Ї–∞ –Є –°–Ъ–§. –Я–Њ –љ–µ–Ї–Њ—В–Њ—А—Л–Љ –і–∞–љ–љ—Л–Љ, –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є –љ–∞—А—Г—И–µ–љ–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї —П–≤–ї—П—О—В—Б—П –±–Њ–ї–µ–µ –≤–∞–ґ–љ—Л–Љ–Є –њ—А–µ–і–Є–Ї—В–Њ—А–∞–Љ–Є —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є, —З–µ–Љ –њ–∞—А–∞–Љ–µ—В—А—Л —Б–µ—А–і–µ—З–љ–Њ–є –і–µ—П—В–µ–ї—М–љ–Њ—Б—В–Є (—Д—А–∞–Ї—Ж–Є—П –≤—Л–±—А–Њ—Б–∞, –Ї–ї–∞—Б—Б –•–°–Э –њ–Њ –Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є–Є –Э—М—О––Щ–Њ—А–Ї—Б–Ї–Њ–є –Р—Б—Б–Њ—Ж–Є–∞—Ж–Є–Є —Б–µ—А–і—Ж–∞) —Г –±–Њ–ї—М–љ—Л—Е –•–°–Э [90].
–° –і—А—Г–≥–Њ–є —Б—В–Њ—А–Њ–љ—Л, –њ–∞—В–Њ–≥–µ–љ–µ–Ј –°–°–Ч —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –•–Я–Э –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є —Б–ї–Њ–ґ–љ—Г—О –Љ–Њ–Ј–∞–Є–Ї—Г. –Ъ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–Љ —Д–∞–Ї—В–Њ—А–∞–Љ–Є —А–Є—Б–Ї–∞, –Њ–±—Л—З–љ—Л–Љ –і–ї—П –Њ–±—Й–µ–є –њ–Њ–њ—Г–ї—П—Ж–Є–Є, –і–Њ–±–∞–≤–ї—П—О—В—Б—П —Д–∞–Ї—В–Њ—А—Л —А–Є—Б–Ї–∞, —В–Є–њ–Є—З–љ—Л–µ –і–ї—П –•–Я–Э. –£ –±–Њ–ї—М–љ—Л—Е —Б –љ–µ—Д—А–Њ–њ–∞—В–Є–µ–є –≤—Л—И–µ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞, —З—В–Њ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ —Н–њ–Є–і–µ–Љ–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—П–Љ–Є (–≤–Њ–Ј—А–∞—Б—В, –љ–∞–ї–Є—З–Є–µ —Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є), —Б–Њ—Ж–Є–∞–ї—М–љ—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤, –∞ —В–∞–Ї–ґ–µ –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–Љ–Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–Љ–Є —Б–Њ–±—Б—В–≤–µ–љ–љ–Њ –•–Я–Э. –Х—Й–µ –±–Њ–ї—М—И–µ —Г–≤–µ–ї–Є—З–Є–≤–∞—О—В —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–є —А–Є—Б–Ї –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–µ –Є –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–µ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –і–ї—П –•–Я–Э (–њ–µ—А–µ–≥—А—Г–Ј–Ї–∞ –Њ–±—К–µ–Љ–Њ–Љ, –∞–љ–µ–Љ–Є—П, –≥–Є–њ–µ—А–њ–∞—А–∞—В–Є—А–µ–Њ–Ј, —Н–ї–µ–Ї—В—А–Њ–ї–Є—В–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П, –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л–є —Б—В—А–µ—Б—Б). –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –љ–µ–Ї–Њ—В–Њ—А—Л–µ –∞–≤—В–Њ—А—Л –њ—А–Њ–≤–Њ–і—П—В –њ–∞—А–∞–ї–ї–µ–ї–Є –Љ–µ–ґ–і—Г –љ–µ—Д—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–Њ–Љ –Є –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–Њ–Љ, –Њ–њ–Є—Б—Л–≤–∞—П –≤–Њ –Љ–љ–Њ–≥–Њ–Љ —Б—Е–Њ–і–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –Є—Е —А–∞–Ј–≤–Є—В–Є—П [91].
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —А–∞–љ–љ–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б–µ—А–і—Ж–∞ (–і–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П) –љ–∞—Е–Њ–і—П—В—Б—П –≤ –љ–∞–Є–±–Њ–ї–µ–µ —Б–Є–ї—М–љ–Њ–є –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є —Б —А–∞–љ–љ–Є–Љ–Є –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є –≤–Њ–≤–ї–µ—З–µ–љ–Є—П –њ–Њ—З–µ–Ї (–Ї–ї—Г–±–Њ—З–Ї–Њ–≤–∞—П –≥–Є–њ–µ—А–њ–µ—А—Д—Г–Ј–Є—П –Є –≥–Є–њ–µ—А—Д–Є–ї—М—В—А–∞—Ж–Є—П, –Ь–Є–Р) –Є –љ–∞—З–∞–ї—М–љ—Л–Љ–Є —Н—В–∞–њ–∞–Љ–Є –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ (—Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П). –Я—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –Є —А–∞–Ј–≤–Є—В–Є–µ –°–°–Ч –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–∞–љ–Њ —Б —Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ –≤–µ—А–Њ—П—В–љ–Њ—Б—В–Є –њ–Њ—П–≤–ї–µ–љ–Є—П –•–Я–Э. –° –і—А—Г–≥–Њ–є —Б—В–Њ—А–Њ–љ—Л, —А–∞–Ј–≤–Є—В–Є–µ –љ–µ—Д—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В —А–Є—Б–Ї —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є –Ј–∞–±–Њ–ї–µ–≤–∞–µ–Љ–Њ—Б—В–Є –Є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є.
–Т—Л–≤–Њ–і—Л –Є –≥–Є–њ–Њ—В–µ–Ј—Л
1. –Я—А–Њ—Б–ї–µ–ґ–Є–≤–∞–µ—В—Б—П –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ —Б—Е–Њ–і—Б—В–≤–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –≤ —Б–µ—А–і—Ж–µ –Є –њ–Њ—З–Ї–∞—Е –≤ –њ—А–Њ—Ж–µ—Б—Б–µ —А–∞–Ј–≤–Є—В–Є—П —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Є –њ–Њ—З–µ—З–љ–Њ–≥–Њ –Ї–Њ–љ—В–Є–љ—Г—Г–Љ–∞.
2. –Т–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є–µ –Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є–є –≤ —Б–µ—А–і—Ж–µ –Є –њ–Њ—З–Ї–∞—Е –≤–Њ –Љ–љ–Њ–≥–Њ–Љ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ —Б—Е–Њ–і–љ—Л–Љ–Є –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–Љ–Є, –љ–µ–є—А–Њ–≥—Г–Љ–Њ—А–∞–ї—М–љ—Л–Љ–Є –Є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–Љ–Є –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞–Љ–Є.
3. –°–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–є –Є –њ–Њ—З–µ—З–љ—Л–є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ —А–∞–Ј–≤–Є–≤–∞—О—В—Б—П –њ–∞—А–∞–ї–ї–µ–ї—М–љ–Њ –Є –≤ —В–µ—Б–љ–Њ–є –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є. –†–∞–љ–љ–Є–µ –Љ–∞—А–Ї–µ—А—Л –≤–Њ–≤–ї–µ—З–µ–љ–Є—П —Б–µ—А–і—Ж–∞ –Ї–Њ—А—А–µ–ї–Є—А—Г—О—В —Б –љ–∞—З–∞–ї—М–љ—Л–Љ–Є –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є –≤–Њ–≤–ї–µ—З–µ–љ–Є—П –њ–Њ—З–µ–Ї, –љ–∞ –±–Њ–ї–µ–µ –њ–Њ–Ј–і–љ–Є—Е —Н—В–∞–њ–∞—Е –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є –њ—А–Њ—Б–ї–µ–ґ–Є–≤–∞—О—В—Б—П –љ–∞ –Є–љ–Њ–Љ —Г—А–Њ–≤–љ–µ – —Г—А–Њ–≤–љ–µ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є –Є –њ–Њ—З–µ—З–љ–Њ–є –Ј–∞–±–Њ–ї–µ–≤–∞–µ–Љ–Њ—Б—В–Є –Є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є. –Ь–µ–ґ–і—Г —В–µ–Љ –љ–µ–ї—М–Ј—П –Њ—В—А–Є—Ж–∞—В—М —Б—Г—Й–µ—Б—В–≤–Њ–≤–∞–љ–Є–µ –Њ–њ—А–µ–і–µ–ї–µ–љ–љ–Њ–є –Є–Ј–±–Є—А–∞—В–µ–ї—М–љ–Њ—Б—В–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П –Њ—А–≥–∞–љ–Њ–≤––Љ–Є—И–µ–љ–µ–є» (–≤ —З–∞—Б—В–љ–Њ—Б—В–Є, «—Б–µ—А–і–µ—З–љ—Л–є», «–њ–Њ—З–µ—З–љ—Л–є», «–Љ–Њ–Ј–≥–Њ–≤–Њ–є» –≤–∞—А–Є–∞–љ—В—Л —В–µ—З–µ–љ–Є—П –Р–У), –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –Ї–Њ—В–Њ—А–Њ–є —В—А–µ–±—Г—О—В –і–∞–ї—М–љ–µ–є—И–µ–≥–Њ –Є–Ј—Г—З–µ–љ–Є—П.




–Ы–Є—В–µ—А–∞—В—Г—А–∞
1. Dzau VJ, Antman EM, Black HR, Hayes DL, Manson JE, Plutzky J, Popma JJ, Stevenson W. The cardiovascular disease continuum validated: clinical evidence of improved patient outcomes: part I: Pathophysiology and clinical trial evidence (risk factors through stable coronary artery disease). Circulation 2006; 114: 2850–2870.
2. Dzau VJ, Antman EM, Black HR, Hayes DL, Manson JE, Plutzky J, Popma JJ, Stevenson W. The cardiovascular disease continuum validated: clinical evidence of improved patient outcomes: part II: Clinical trial evidence (acute coronary syndromes through renal disease) and future directions. Circulation 2006; 114: 2871–2891.
3. –Я–Њ–і–Ј–Њ–ї–Ї–Њ–≤–∞ –Э–Ь, –Я–Њ–і–Ј–Њ–ї–Ї–Њ–≤ –Т–Ш, –Ь–Њ–ґ–∞—А–Њ–≤–∞ –Ы–У, –•–Њ–Љ–Є—Ж–Ї–∞—П –Ѓ–Т. –У–Њ—А–Љ–Њ–љ–∞–ї—М–љ—Л–є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ –ґ–µ–љ—Б–Ї–Њ–≥–Њ –Ј–і–Њ—А–Њ–≤—М—П: —Н–≤–Њ–ї—О—Ж–Є—П —Б–µ—А–і–µ—З–љ–Њ–c–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ —А–Є—Б–Ї–∞ –Њ—В –Љ–µ–љ–∞—А—Е–µ –і–Њ –Љ–µ–љ–Њ–њ–∞—Г–Ј—Л. –°–µ—А–і—Ж–µ 2004; 3: 20–24.
4. –С–µ–ї–µ–љ–Ї–Њ–≤ –Ѓ–Э, –Ь–∞—А–µ–µ–≤ –Т–Ѓ. –°–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В—Л–є –Ї–Њ–љ—В–Є–љ—Г—Г–Љ. –°–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М 2002; 3: 7–11.
5. Phillips RA, Goldman ME, Ardeljan M, Arora R, Eison HB, Yu BY, Krakoff LR. Determinants of abnormal left ventricular filling in early hypertension. J Am Coll Cardiol 1989; 14: 979–985.
6. Mayet J, Foale RA. Changes in left ventricular function in cardiac hypertrophy. In Sheridan DJ (ed) Left ventricular hypertrophy, Edition London, UK: Churchill Ltd 1998; 93–99.
7. Phillips RA, Diamond JA. Left ventricular hypertrophy, congestive heart failure, and coronary flow reserve abnormalities in hypertension. In Oparil S, Weber MA (eds): Hypertension: a companion to Brenner & Rector’s The Kidney, Edition Philadelphia, Pennsylvania, US: WB Saunders Company 2000; 244–277.
8. –Р–≥–µ–µ–≤ –§–Ґ, –Ю–≤—З–Є–љ–љ–Є–Ї–Њ–≤ –Р–У. –Ф–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Ї–∞–Ї –њ—А–Њ—П–≤–ї–µ–љ–Є–µ —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є—П —Б–µ—А–і—Ж–∞. –°–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М 2002; 3: 190–195.
9. –®–ї—П—Е—В–Њ –Х–Т, –®–≤–∞—А—Ж –Х–Ш, –Э–µ—Д–µ–і–Њ–≤–∞ –Ѓ–С, –Ъ–Њ–љ—А–∞–і–Є –Р–Ю, –†—Г–і–Њ–Љ–∞–љ–Њ–≤, –Ю.–У., –Т–Є–љ–љ–Є–Ї –Ґ–Р. –Ф–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П —Г –±–Њ–ї—М–љ—Л—Е –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ—М—О: —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М, –≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–µ, –і–µ–Љ–Њ–≥—А–∞—Д–Є—З–µ—Б–Ї–Є–µ –Є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –і–µ—В–µ—А–Љ–Є–љ–∞–љ—В—Л. –°–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М 2003; 4: 187–189.
10. Banerjee P, Clark AL, Nikitin N, Cleland JG. Diastolic heart failure. Paroxysmal or chronic? Eur J Heart Fail 2004; 6: 427–431.
11. Grossman W. Defining diastolic dysfunction. Circulation 2000; 101: 2020–2021.
12. 01Belenkov, Aaeaieia I. ?aiiaaee?iaaiea eaaiai ?aeoai?ea: eiiieaeniue iiaoia. Na?aa?iay iaainoaoi?iinou 2002; 3: 161–163.
13. McMurray JJV, Stewart S. The burden of heart failure. Eur Heart J Suppl 2003; 5: I3–I13.
14. Kannel WB. Prevalence and natural history of electrocardiographic left ventricular hypertrophy. Am J Med 1983; 75: 4–11.
15. de Wardener HE. The primary role of the kidney and salt intake in the aetiology of essential hypertension: Part I. Clin Sci (Lond) 1990; 79: 193–200.
16. Ruilope LM, Campo C, Rodriguez–Artalejo F, Lahera V, Garcia–Robles R, Rodicio JL. Blood pressure and renal function: therapeutic implications. J Hypertens 1996; 14: 1259–1263.
17. USRDS: U.S. Renal Data System. USRDS 2002 Annual Data Report: Atlas of End–Stage Renal Disease in the nited States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2002. In Edition.
18. Meyrier A, Simon P. Nephroangiosclerosis and hypertension: things are not as simple as you might think. Nephrol Dial Transplant 1996; 11: 2116–2120.
19. Ritz E, Orth S, Weinreich T, Wagner J. Systemic hypertension versus intraglomerular hypertension in progression. Kidney Int 1994; 45: 438–442.
20. Weisstuch JM, Dworkin LD. Does essential hypertension cause end–stage renal disease? Kidney Int Suppl 1992; 36: S33–37.
21. Luke RG. Hypertensive nephrosclerosis: pathogenesis and prevalence. Essential hypertension is an important cause of end–stage renal disease. Nephrol Dial Transplant 1999; 14: 2271–2278.
22. Herrera–Acosta J. The role of systemic and glomerular hypertension in progressive glomerular injury. Kidney Int Suppl 1994; 45: S6–10.
23. Bulatov VA, Stenehjem A, Os I. Left ventricular mass assessed by electrocardiography and albumin excretion rate as a continuum in untreated essential hypertension. J Hypertens 2001; 19: 1473–1478.
24. Redon J, Williams B. Microalbuminuria in essential hypertension: redefining the threshold. J Hypertens 2002; 20: 353–355.
25. Ruilope LM. Kidney dysfunction: a sensitive predictor of cardiovascular risk. Am J Hypertens 2001; 14: 213S–217S.
26. Adler CP. Polyploidization and augmentation of heart muscle cells during normal cardiac growth and in cardiac hypertrophy. In Oberpriller JO, Oberpriller JC, Mauro A (eds): The development and regenerative potential of cardiac muscle, Edition Chur: Harwood Academic Publishers 1991; 227–252.
27. Weber KT, Sun Y, Dhalla AK, R.V. G. Extracellular matrix and fibrosis in cardiac hypertrophy. In Sheridan DJ (ed) Left Ventricular Hypertrophy, Edition 1. London, UK: Churchill Ltd 1998; 37–44.
28. Sheridan DJ, Kingsbury MP. Mechanisms od reduced coronary reserve in cardiac hypertrophy. In Sheridan DJ (ed) Left ventricular hypertrophy, Edition London, UK: Churchill Communication Europe Ltd 1998; 135–143.
29. Strauer BE. Significance of coronary circulation in hypertensive heart disease for development and prevention of heart failure. Am J Cardiol 1990; 65: 34G–41G.
30. Levy BI, Ambrosio G, Pries AR, Struijker–Boudier HA. Microcirculation in hypertension: a new target for treatment? Circulation 2001; 104: 735–740.
31. Rizzoni D, Castellano M, Porteri E, Bettoni G, Muiesan ML, Cinelli A, Rosei EA. Effects of low and high doses of fosinopril on the structure and function of resistance arteries. Hypertension 1995; 26: 118–123.
32. Schlessinger SD, Tankersley MR, Curtis JJ. Clinical documentation of end–stage renal disease due to hypertension. Am J Kidney Dis 1994; 23: 655–660.
33. Zarif L, Covic A, Iyengar S, Sehgal AR, Sedor JR, Schelling JR. Inaccuracy of clinical phenotyping parameters for hypertensive nephrosclerosis. Nephrol Dial Transplant 2000; 15: 1801–1807.
34. Caetano ER, Zatz R, Saldanha LB, Praxedes JN. Hypertensive nephrosclerosis as a relevant cause of chronic renal failure. Hypertension 2001; 38: 171–176.
35. Tracy RE, Ishii T. What is ‘nephrosclerosis’? lessons from the US, Japan, and Mexico. Nephrol Dial Transplant 2000; 15: 1357–1366.
36. Komuro I, Kaida T, Shibazaki Y, Kurabayashi M, Katoh Y, Hoh E, Takaku F, Yazaki Y. Stretching cardiac myocytes stimulates protooncogene expression. J Biol Chem 1990; 265: 3595–3598.
37. Komuro I, Katoh Y, Kaida T, Shibazaki Y, Kurabayashi M, Hoh E, Takaku F, Yazaki Y. Mechanical loading stimulates cell hypertrophy and specific gene expression in cultured rat cardiac myocytes. Possible role of protein kinase C activation. J Biol Chem 1991; 266: 1265–1273.
38. Yamazaki T, Komuro I, Yazaki Y. Triggers for cardiac hypertrophy. In Sheridan DJ (ed) Left ventricular hypertrophy, Edition 1. London, UK: Churchill Ltd 1998; 71–76.
39. Baker KM, Booz GW, Dostal DE. Cardiac actions of angiotensin II: Role of an intracardiac renin–angiotensin system. Annu Rev Physiol 1992; 54: 227–241.
40. Schunkert H, Hense HW, Muscholl M, Luchner A, Kurzinger S, Danser AH, Riegger GA. Associations between circulating components of the renin–angiotensin–aldosterone system and left ventricular mass. Heart 1997; 77: 24–31.
41. Agabiti–Rosei E, Muiesan ML. Hypertensive left ventricular hypertrophy: pathophysiological and clinical issues. Blood Press 2001; 10: 288–298.
42. Paul M, Ganten D. The molecular basis of cardiovascular hypertrophy: the role of the renin–angiotensin system. J Cardiovasc Pharmacol 1992; 19 Suppl 5: S51–58.
43. Schlaich MP, Schobel HP, Hilgers K, Schmieder RE. Impact of aldosterone on left ventricular structure and function in young normotensive and mildly hypertensive subjects. Am J Cardiol 2000; 85: 1199–1206.
44. Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin–angiotensin–aldosterone system. Circulation 1991; 83: 1849–1865.
45. Sen S, Tarazi RC, Khairallah PA, Bumpus FM. Cardiac hypertrophy in spontaneously hypertensive rats. Circ Res 1974; 35: 775–781.
46. Laks MM, Morady F, Swan HJ. Myocardial hypertrophy produced by chronic infusion of subhypertensive doses of norepinephrine in the dog. Chest 1973; 64: 75–78.
47. Devereux RB, Pickering TG, Cody RJ, Laragh JH. Relation of renin–angiotensin system activity to left ventricular hypertrophy and function in experimental and human hypertension. J Clin Hypertens 1987; 3: 87–103.
48. –®–ї—П—Е—В–Њ –Х–Т, –Ъ–Њ–љ—А–∞–і–Є –Р–Ю. –†–Њ–ї—М –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤ –≤ —А–µ–Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є–Є —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–Є—Б—В–µ–Љ—Л –њ—А–Є –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є. –Р—А—В–µ—А–Є–∞–ї—М–љ–∞—П –≥–Є–њ–µ—А—В–µ–љ–Ј–Є—П 2002; 8: 107–114.
49. Post WS, Larson MG, Myers RH, Galderisi M, Levy D. Heritability of left ventricular mass: the Framingham Heart Study. Hypertension 1997; 30: 1025–1033.
50. Staessen JA, Wang JG, Ginocchio G, Petrov V, Saavedra AP, Soubrier F, Vlietinck R, Fagard R. The deletion/insertion polymorphism of the angiotensin converting enzyme gene and cardiovascular–renal risk. J Hypertens 1997; 15: 1579–1671.
51. Alvestrand A, Gutierrez A, Bucht H, Bergstrom J. Reduction of blood pressure retards the progression of chronic renal failure in man. Nephrol Dial Transplant 1988; 3: 624–631.
52. Brazy PC, Stead WW, Fitzwilliam JF. Progression of renal insufficiency: role of blood pressure. Kidney Int 1989; 35: 670–674.
53. Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Brenner BM. Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol 1981; 241: F85–93.
54. Brenner BM, Meyer TW, Hostetter TH. Dietary protein intake and the progressive nature of kidney disease: the role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N Engl J Med 1982; 307: 652–659.
55. Dirks JH, Brenner BM. Mechanisms of injury in progressive renal disease: Insights from experimental daya. Kidney Int 1994; 45: S–22–S–23.
56. Sharma AM. Renal involvement in hypertensive cardiovascular disease. Eur Heart J 2003; 5: F12–F18.
57. Rodriguez–Iturbe B, Pons H, Herrera–Acosta J, Johnson RJ. Role of immunocompetent cells in nonimmune renal diseases. Kidney Int 2001; 59: 1626–1640.
58. Leehey DJ, Singh AK, Alavi N, Singh R. Role of angiotensin II in diabetic nephropathy. Kidney Int Suppl 2000; 77: S93–98.
59. Hesse IF, Johns EJ. The subtype of alpha–adrenoceptor involved in the neural control of renal tubular sodium reabsorption in the rabbit. J Physiol 1984; 352: 527–538.
60. Osborn JL, Holdaas H, Thames MD, DiBona GF. Renal adrenoceptor mediation of antinatriuretic and renin secretion responses to low frequency renal nerve stimulation in the dog. Circ Res 1983; 53: 298–305.
61. DiBona GF. Neural control of renal function: role of renal alpha adrenoceptors. J Cardiovasc Pharmacol 1985; 7 Suppl 8: S18–23.
62. Iglesias J, Levine JS. Albuminuria and renal injury—beware of proteins bearing gifts. Nephrol Dial Transplant 2001; 16: 215–218.
63. Williams JD, Coles GA. Proteinuria—a direct cause of renal morbidity? Kidney Int 1994; 45: 443–450.
64. Keane WF. Lipids and the kidney. Kidney Int 1994; 46: 910–920.
65. Bianchi S, Bigazzi R, Campese VM. Microalbuminuria in essential hypertension: significance, pathophysiology, and therapeutic implications. Am J Kidney Dis 1999; 34: 973–995.
66. van Hooft IM, Grobbee DE, Derkx FH, de Leeuw PW, Schalekamp MA, Hofman A. Renal hemodynamics and the renin–angiotensin–aldosterone system in normotensive subjects with hypertensive and normotensive parents. N Engl J Med 1991; 324: 1305–1311.
67. Samuelsson O, Attman PO, Larsson R, Mulec H, Rymo L, Weiss L, Ricksten A. Angiotensin I–converting enzyme gene polymorphism in non–diabetic renal disease. Nephrol Dial Transplant 2000; 15: 481–486.
68. Mallamaci F, Zuccala A, Zoccali C, Testa A, Gaggi R, Spoto B, Martorano C, Curatola A, Misefari V, Cuzzola F, Romeo G, Zucchelli P. The deletion polymorphism of the angiotensin–converting enzyme is associated with nephroangiosclerosis. Am J Hypertens 2000; 13: 433–437.
69. Fernandez–Llama P, Poch E, Oriola J, Botey A, Coll E, Darnell A, Rivera F, Revert L. Angiotensin converting enzyme gene I/D polymorphism in essential hypertension and nephroangiosclerosis. Kidney Int 1998; 53: 1743–1747.
70. Gumprecht J, Zychma MJ, Grzeszczak W, Zukowska–Szczechowska E. Angiotensin I–converting enzyme gene insertion/deletion and angiotensinogen M235T polymorphisms: risk of chronic renal failure. End–Stage Renal Disease Study Group. Kidney Int 2000; 58: 513–519.
71. Coll E, Campos B, Gonzalez–Nunez D, Botey A, Poch E. Association between the A1166C polymorphism of the angiotensin II receptor type 1 and progression of chronic renal insufficiency. J Nephrol 2003; 16: 357–364.
72. Silberberg JS, Barre PE, Prichard SS, Sniderman AD. Impact of left ventricular hypertrophy on survival in end–stage renal disease. Kidney Int 1989; 36: 286–290.
73. Randomised placebo–controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non–diabetic nephropathy. The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Lancet 1997; 349: 1857–1863.
74. Ruggenenti P, Perna A, Mosconi L, Pisoni R, Remuzzi G. Urinary protein excretion rate is the best independent predictor of ESRF in non–diabetic proteinuric chronic nephropathies. «Gruppo Italiano di Studi Epidemiologici in Nefrologia» (GISEN). Kidney Int 1998; 53: 1209–1216.
75. Brown MJ, Palmer CR, Castaigne A, de Leeuw PW, Mancia G, Rosenthal T, Ruilope LM. Morbidity and mortality in patients randomised to double–blind treatment with a long–acting calcium–channel blocker or diuretic in the International Nifedipine GITS study: Intervention as a Goal in Hypertension Treatment (INSIGHT). Lancet 2000; 356: 366–372.
76. Kannel WB, Stampfer MJ, Castelli WP, Verter J. The prognostic significance of proteinuria: the Framingham study. Am Heart J 1984; 108: 1347–1352.
77. Zanchetti A, Hansson L, Dahlof B, Elmfeldt D, Kjeldsen S, Kolloch R, Larochelle P, McInnes GT, Mallion JM, Ruilope L, Wedel H. Effects of individual risk factors on the incidence of cardiovascular events in the treated hypertensive patients of the Hypertension Optimal Treatment Study. HOT Study Group. J Hypertens 2001; 19: 1149–1159.
78. Campese VM, Bianchi S, Bigazzi R. Is microalbuminuria a predictor of cardiovascular and renal disease in patients with essential hypertension? Curr Opin Nephrol Hypertens 2000; 9: 143–147.
79. Ruilope LM. The kidney as a sensor of cardiovascular risk in essential hypertension. J Am Soc Nephrol 2002; 13 Suppl 3: S165–168.
80. Viberti GC, Hill RD, Jarrett RJ, Argyropoulos A, Mahmud U, Keen H. Microalbuminuria as a predictor of clinical nephropathy in insulin–dependent diabetes mellitus. Lancet 1982; 1: 1430–1432.
81. Bigazzi R, Bianchi S, Baldari D, Campese VM. Microalbuminuria predicts cardiovascular events and renal insufficiency in patients with essential hypertension. J Hypertens 1998; 16: 1325–1333.
82. Erley CM, Risler T. Microalbuminuria in primary hypertension: is it a marker of glomerular damage? Nephrol Dial Transplant 1994; 9: 1713–1715.
83. Bianchi S, Bigazzi R, Campese VM. Microalbuminuria in essential hypertension: significance, pathophysiology, and therapeutic implications. Am J Kidney Dis 1999; 34: 973–995.
84. Agewall S, Wikstrand J, Ljungman S, Herlitz H, Fagerberg B. Does microalbuminuria predict cardiovascular events in nondiabetic men with treated hypertension? Risk Factor Intervention Study Group. Am J Hypertens 1995; 8: 337–342.
85. Pedrinelli R. Microalbuminuria in essential hypertension. A marker of systemic vascular damage? Nephrol Dial Transplant 1997; 12: 379–381.
86. Dries DL, Exner DV, Domanski MJ, Greenberg B, Stevenson LW. The prognostic implications of renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol 2000; 35: 681–689.
87. Schrier RW, Abraham WT. Hormones and hemodynamics in heart failure. N Engl J Med 1999; 341: 577–585.
88. Magri P, Rao MA, Cangianiello S, Bellizzi V, Russo R, Mele AF, Andreucci M, Memoli B, De Nicola L, Volpe M. Early impairment of renal hemodynamic reserve in patients with asymptomatic heart failure is restored by angiotensin II antagonism. Circulation 1998; 98: 2849–2854.
89. Volpe M, Magri P, Rao MA, Cangianiello S, DeNicola L, Mele AF, Memoli B, Enea I, Rubattu S, Gigante B, Trimarco B, Epstein M, Condorelli M. Intrarenal determinants of sodium retention in mild heart failure: effects of angiotensin–converting enzyme inhibition. Hypertension 1997; 30: 168–176.
90. Hillege HL, Girbes AR, de Kam PJ, Boomsma F, de Zeeuw D, Charlesworth A, Hampton JR, van Veldhuisen DJ. Renal function, neurohormonal activation, and survival in patients with chronic heart failure. Circulation 2000; 102: 203–210.
91. Locatelli F, Marcelli D, Conte F, D’Amico M, Del Vecchio L, Limido A, Malberti F, Spotti D. Cardiovascular disease in chronic renal failure: the challenge continues. Registro Lombardo Dialisi e Trapianto. Nephrol Dial Transplant 2000; 15 Suppl 5: 69–80.



