




Сердечная недостаточность (СН) представляет собой огромную медико–социальную проблему [1,2]. СН относится к распространенным заболеваниям – ею страдает более 2% жителей США (почти 5 млн. человек), а в течение года после постановки диагноза умирают 30–40% пациентов [3]. СН является инвалидизирующей патологией, значительно снижая качество жизни больного. На нее тратится около 2% бюджета Государственной службы здравоохранения Великобритании, а в США на лечение СН ежегодно расходуется около 28 млрд. долларов. Более того, затраты на СН в последующее десятилетие будут только возрастать из–за старения населения и разработки новых методов лечения причин СН.
Внедрение в клиническую практику ингибиторов ангиотензинпревращающего фермента (АПФ) [4,5], антагонистов альдостерона [6], блокаторов b–адренорецепторов [7,8] и методов ресинхронизационной терапии [9,10] позволило добиться существенного прогресса в лечении хронической СН за последние 20 лет. Однако несмотря на наличие даже самых современных медицинских технологий, показатели ежегодной смертности от СН составляют примерно 10% [10]. Поэтому поиск новых методов лечения СН представляется одной из основных задач кардиологии.
Хроническая СН многофакторна. Существует множество причин перебоев сердечной деятельности у человека [11], однако имеющиеся данные позволяют предположить, что плохо работающее сердце – это своего рода «двигатель без топлива», то есть не исключено, что важное звено в патогенезе СН составляет нарушение энергообмена в миокарде. Следовательно, модуляция метаболических процессов в сердечной мышце может стать новым направлением в разработке подходов к лечению СН.
В данном обзоре представлены данные о механизмах энергообмена в миокарде и методы его изучения, проанализирована роль нарушений энергообмена в развитии СН, описаны различные варианты метаболической терапии.
Гипотеза «энергетического
голодания»
Сердце при СН образно сравнивают с двигателем, лишенным энергии и топлива. Такая концепция не нова: еще в 1939 г. о значительном снижении содержания креатина в миокарде при СН писали Hermann и Decherd в статье «Химическая природа сердечной недостаточности» [12]. В дальнейшем теорию о недостаточном поступлении энергии разрабатывали различные научные группы [13–15], а процессы энергообмена в сердечной мышце остаются объектом пристального внимания ученых и сегодня [16–24]. Это становится особенно актуальным в свете того, что использование в лечении СН лекарственных препаратов, позволяющих сэкономить энергетические ресурсы, таких как блокаторы b–адренорецепторов [7,8], ингибиторы АПФ [4,5] или антагонисты ангиотензина II [25,26], заметно улучшает прогноз заболевания. Про СН еще говорят так: «Сердце – это ослабевшая, уставшая лошадь, но если ее хорошенько накормить, она снова сможет работать, хотя и не так интенсивно, как раньше» [27].
Процессы энергообмена в миокарде
Недостаток энергии в миокарде – основная причина СН [18]. Сердце потребляет больше энергии, чем какой–либо другой орган. На его работу затрачивается около 6 кг АТФ в день, что в 20–30 раз больше его собственного веса. Ежедневно сердце совершает примерно 100000 сократительных движений, перекачивая более 10 т крови. Для получения энергии, необходимой для поддержания сердечной деятельности, в миокарде функционирует механизм превращения энергии химических связей молекул жирных кислот и глюкозы в механическую энергию взаимодействия актина и миозина, из которых построены миофибриллы. Нарушение способности генерировать достаточное количество энергии обусловливает возникновение механических перебоев в работе сердца.
Звенья энергообмена в миокарде
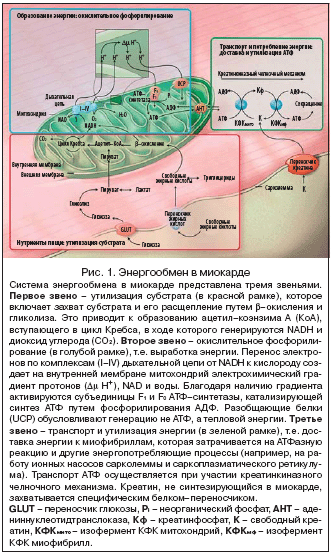
Процессы энергообмена в сердечной мышце комплексны (рис. 1). Существует три ключевых звена этой системы. Первое звено – утилизация субстрата, т.е. нутриентов пищи. При этом свободные жирные кислоты и глюкоза захватываются клетками, расщепляются путем b–окисления и гликолиза, а образовавшиеся промежуточные метаболиты вступают в цикл Кребса. Второе звено энергообмена – выработка энергии в реакциях окислительного фосфорилирования на уровне дыхательной цепи митохондрий. В результате фосфорилирования АДФ синтезируется высокоэнергетический АТФ, который служит непосредственным источником энергии для всех энергопотребляющих процессов в миокарде. Наконец, третье звено энергообмена – транспорт и утилизация АТФ миофибриллами. Этот процесс обусловливает перенос энергии при участии так называемого креатинкиназного челночного механизма.
Креатинкиназная система
Являясь одним из компонентов третьего звена энергообмена в сердечной мышце (транспорт и утилизация АТФ), креатинфосфокиназа (КФК) переносит энергию фосфатных связей АТФ на креатин с образованием креатинфосфата (Кф) и АДФ. Кф, будучи по размеру меньше АТФ, быстро поступает из митохондрий к миофибриллам, КФК которых катализирует ресинтез АТФ из Кф. Свободный креатин, генерируемый в результате отщепления фосфатной группы от Кф, транспортируется обратно в митохондрии.
Креатин синтезируется в печени и в почках, а затем доставляется в миокард, где захватывается специфическим мембранным белком–переносчиком креатина [31] против 50–кратного градиента концентрации. В миокарде КФК катализирует фосфорилирование примерно 2/3 общего пула креатина с образованием Кф, а оставшаяся 1/3 циркулирует в форме свободного креатина. Небольшое количество креатина постоянно теряется за счет пассивной диффузии через сарколемму [32]. Ключевая роль системы КФК состоит в том, что она функционирует в качестве энергетического буфера. Когда энергозатраты превосходят энергообеспечение, уровень Кф снижается, что позволяет поддерживать нормальное содержание АТФ, но концентрация свободного АДФ при этом растет [29]. При высокой концентрации свободного АДФ активность большинства внутриклеточных ферментов подавляется, что влечет за собой нарушение сократительной деятельности сердца. Таким образом, если уровень Кф падает, а концентрация свободного АДФ повышается, то даже на фоне неизмененного содержания АТФ в кардиомиоцитах возникают расстройства метаболизма.
Методы оценки
энергообмена в миокарде
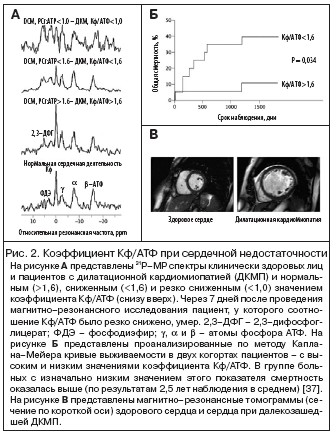
Функционирование различных звеньев системы энергоснабжения сердечной мышцы можно оценить стандартными методами в биоптатах миокарда, которые получают при биопсии, в процессе трансплантации или в ходе экспериментов на подопытных животных. Однако проанализировать, например, содержание АТФ и Кф в образцах тканей проблематично из–за нестабильности этих молекул [29]. По этой причине для определения уровней АТФ и Кф используют магнитно–резонансную спектроскопию с фосфором–31 (31Р–МР) [33–36]. У подопытных грызунов ее проводят в режиме сильного магнитного поля (до 12 тесла; тесла – единица измерения силы магнитного поля), а у человека – в режиме стандартной магнитно–резонансной томографии (МРТ) при силе магнитного поля до 1,5 тесла. Как видно из рисунка 2, пики на карте спектров 31Р–МР – это сигналы от Кф и трех атомов фосфора АТФ (g–АТФ, a–АТФ и b–АТФ), пропорциональные концентрации этих соединений. МРТ позволяет получить серию последовательных изображений сердца в определенный момент времени и на основе этого подсчитать значения показателей функций миокарда. Наиболее информативным методом изучения процессов энергообмена в сердечной мышце при СН служит определение коэффициента оборачиваемости глюкозы и жирных кислот [38–41], а также скорости окислительного фосфорилирования [42] и транспорта АТФ [34,36]. С методологической точки зрения необходимо учитывать такой аспект, как компартментализация внутриклеточного пространства [43]. Измеряя содержание АТФ, Кф или АДФ внутри клетки в среднем, нельзя судить о том, нормально или нет функционирует кардиомиоцит; для этого следует вычислить концентрацию этих молекул в пространстве, окружающем миофибриллы, в области саркоплазматического ретикулума и ионных насосов сарколеммы. Однако методов для проведения таких измерений пока нет, и поэтому их приходится экстраполировать из общих оценок.
Нарушение энергообмена при СН
Изменения энергообмена в сердечной мышце при СН представлены на рисунке 3. Эти данные были получены как в опытах на животных [19,44–57], так и из результатов клинических наблюдений [58–68]. Указанные изменения затрагивают все три звена энергообеспечения миокарда – утилизацию субстрата, окислительное фосфорилирование и метаболизм макроэргических фосфатов.
Утилизация субстрата
Расстройство процессов утилизации субстрата, ведущее к перебоям в работе сердца при СН, может быть обусловлено нарушением захвата субстрата, его окисления или тем и другим. Кроме того, расстройство процессов утилизации субстрата может возникать и вследствие изменения соотношения жирных кислот (60–90%) и глюкозы (10–40%), используемых в синтезе АТФ. Хотя данные исследований, посвященных изучению процессов утилизации субстрата при СН, весьма противоречивы, большинство из них все же свидетельствует о том, что на ранних стадиях СН утилизация жирных кислот не меняется или незначительно усиливается [19,44], однако при выраженной СН активность этого процесса резко падает [45]. Данные по метаболизму глюкозы также неоднозначны, но многими исследованиями доказано, что уже на ранних стадиях СН его интенсивность начинает нарастать [46,47]. При прогрессировании СН кардиомиоциты приобретают резистентность к инсулину и утилизация глюкозы нарушается. Но эти данные достаточно сложно интерпретировать, поскольку при СН в сыворотке крови повышается содержание свободных жирных кислот, глюкозы и инсулина. Из–за этого отдифференцировать истинные нарушения обмена веществ в сердечной мышце от изменений, отражающих общий метаболический дисбаланс в организме, достаточно сложно [19].
Окислительное фосфорилирование
Расстройство процессов окислительного фосфорилирования может вызвать ухудшение работы кардиомиоцитов вследствие недостаточного поступления АТФ к ним. При СН выявляются структурные аномалии митохондрий кардиомиоцитов; кроме того, увеличивается и количество митохондрий [49]. Одновременно снижается активность цепи переноса электронов и АТФ–синтетазы [50,61,69]; нарушаются процессы окислительного фосфорилирования, опосредованные акцепторами фосфатных групп АДФ, АТФ и креатином [41]; и возрастает содержание разобщающих белков (что обусловливает генерацию митохондриями тепловой энергии, а не АТФ) [70]. Все это приводит к значительному ограничению потребления кислорода и уменьшению выработки энергии в миокарде.
Метаболизм
макроэргических фосфатов
Нарушение транспорта и утилизации АТФ вызывает ухудшение сократительной активности кардиомиоцитов за счет падения средней концентрации АТФ, ослабления транспорта АТФ через КФК (таким образом, что энергия макроэргических фосфатных связей переносится из митохондрий на миофибриллы) или повышения уровня свободного АДФ.
На ранних стадиях СН содержание АТФ в миокарде остается нормальным (порядка 10 ммоль/л), а по мере ее прогрессирования оно снижается на 30–40% [65,66,71]. При этом средняя концентрация АТФ гораздо выше той, которая необходима для реализации процессов, требующих затрат АТФ, например, взаимодействия миозина с АТФазой, т.е. этот фактор не ограничивает работоспособность сердца при СН. Однако уровень Кф и общего креатина начинает снижаться раньше и в большей степени (на 30–70%) [66,67]. Подавление активности белка–переносчика креатина при СН влечет за собой падение уровней общего креатина и, как следствие, Кф [72,73].
СН сопровождается выраженными изменениями функций креатинкиназной системы [67,74–78]. Активность митохондриальной КФК может снижаться до 20% от нормы, а миофибриллярной КФК – до 50%. Отток макроэргических фосфатов и ослабление функций КФК существенно тормозит транспорт АТФ [53,54,79,80], препятствуя тем самым поступлению энергии в клетку и ограничивая ее доставку к миофибриллам на 71% [81]. Описанные нарушения энергообмена могут спровоцировать ухудшение сократительной способности сердечной мышцы и частично потерю инотропного ресурса кардиомиоцитов, что весьма характерно для СН.
При стимуляции катехоламинами, которые создают режим высокой рабочей нагрузки, концентрация свободного АДФ в миокарде возрастает примерно в 2 раза по сравнению с нормой [82]. В условиях такой высокой нагрузки растущая концентрация свободного АДФ в пространстве, окружающем миофибриллы, а также в микрокомпартментах вокруг саркоплазматического ретикулума и ионных насосов сарколеммы уменьшает сократительный ресурс миокарда, клиническим проявлением чего служит развитие одышки при физической нагрузке.
Многие данные, касающиеся перестройки процессов энергообмена в миокарде при СН у человека, были получены при проведении 31Р–МР–спектроскопии. Этот метод можно использовать для вычисления соотношения Кф к АТФ – информативного показателя, отражающего состояние энергообмена в сердечной мышце. При значении этого коэффициента ~100 равновесие креатинкиназной реакции сдвигается в сторону синтеза АТФ из Кф. Следовательно, когда потребление энергии превышает возможности ее образования, то вначале падает содержание Кф, и лишь когда его запасы оказываются исчерпаны, начинает расходоваться АТФ. Однако при СН подключается дополнительный механизм: общий уровень креатина снижается и соотношение Кф/АТФ тоже начинает уменьшаться (рис. 2) [62,63,68]. Степень снижения коэффициента Кф/АТФ в миокарде коррелирует с функциональным классом СН по классификации Нью–Йоркской кардиологической ассоциации [63], с одной стороны, и значениями показателей работы сердца в систолу [83] и в диастолу [84], с другой. Согласно результатам исследования, проведенного у 39 пациентов с дилатационной кардиомиопатией, коэффициент Кф/АТФ может рассматриваться как более информативный по сравнению с остальными клиническими и функциональными показателями прогностический критерий (рис. 2Б) [37], хотя эти результаты требуют дальнейшего подтверждения.
Гипертрофическая кардиомиопатия – наглядный пример дефицита энергии в сердечной мышце [85]. При гипертрофической кардиомиопатии вне зависимости от наличия или отсутствия гипертрофии левого желудочка соотношение Кф/АТФ в миокарде снижается, что обусловлено множественными специфическими аномалиями саркомера [86]. Поскольку нарушение энергообмена происходит уже на ранних стадиях первичных заболеваний миокарда, не исключено, что оно служит и триггером гипертрофической кардиомиопатии.
Регуляция энергообмена
на молекулярном уровне
Количество энергии, расходуемой сердцем, варьирует в широких пределах в зависимости от стадии эмбрионального развития, а также меняется в условиях физиологического и патологического стресса. Выработка энергии тесно взаимосвязана с ее потреблением, однако сердечная мышца практически лишена способности запасать субстрат. Тем не менее существуют механизмы, которые индуцируют экспрессию генов, кодирующих структуру молекул–регуляторов энергообмена [87].
Транскрипционные
факторы рецепторов ядра
Производные метаболизма липидов активируют различные транскрипционные факторы–ядерные рецепторы по механизму, сходному с таковым для стероидных гормонов. Эти транскрипционные факторы быстро запускают экспрессию генов с изменением профиля субстратов, а для функционирования этих факторов обычно требуются коактиваторные белки. Среди таких факторов лучше всего изучены ядерные рецепторы для белков семейства пероксисомных пролифератор–активируемых рецепторов (PRAR), которые представлены тремя изоформами – a, b и g. Все три изоформы способны воздействовать на метаболизм липидов в сердечной мышце, но в наибольшей степени это характерно для PRARa, который контролирует экспрессию ферментов, непосредственно участвующих в окислении жирных кислот. При гипертрофии миокарда как у подопытных животных [88], так и у человека [89,90] экспрессия PRARa ослабляется пропорционально угнетению процессов утилизации жирных кислот. Приведенные данные служат основанием для предположения, что подавление экспрессии PRARa представляет собой ключевой механизм, опосредующий «переключение» утилизации субстратов с жирных кислот на глюкозу, типичное для гипертрофии миокарда.
Ядерный рецептор–коактиватор, коактиватор–1 PRARg (или PCG–1a) – это ключевой регулятор метаболизма митохондрий. Он активирует множество генов, которые отвечают за захват и окисление жирных кислот и за окислительное фосфорилирование [87]. Из этих генов следует отметить PRARa и PRARb, а также ядерные респираторные факторы 1 и 2. Экспериментальные данные свидетельствуют о том, что блокирование PCG–1a [91,92], которое может быть прямым следствием высокого содержания в крови катехоламинов [93], приводит к подавлению экспрессии митохондриальных генов [20]. Это, в свою очередь, способствует нарушению процессов окислительного фосфорилирования в миокарде при СН. Дефицит PCG–1a ускоряет прогрессирование СН [93], а следовательно, этот коактиватор может обладать кардиопротекторным действием.
Несмотря на достигнутые успехи, требуется провести еще целую серию дополнительных исследований, которые помогут выяснить, носят ли изменения метаболических сигналов адаптивный или дезадаптивный характер, или же в зависимости от стадии СН справедливо и то и другое. Регуляторы процессов транспорта креатина и экспрессии КФК на молекулярном уровне пока не идентифицированы.
Экспериментальные животные с «выбитыми» генами и мутациями с потерей функций
Роль нарушений энергообеспечения сердечной мышцы в патогенезе СН многие десятилетия подвергалась сомнению, и существующие противоречия до сих пор не разрешены. Один из возможных вариантов решения проблемы – проведение исследований на мышах с селективным «нокаутом» (потерей функций) генов, отвечающих за метаболические реакции, или изучение врожденных аномалий метаболизма у человека, вызванных мутацией одного гена. Делеция различных генов, которые кодируют специфические факторы метаболизма, участвующие в утилизации субстрата, окислительном фосфорилировании и метаболизме макроэргических фосфатов, уменьшает сократительные резервы сердечной мышцы, способствуетманифестации СН и гипертрофии миокарда, вызывает тахиаритмию или брадиаритмию. Результаты генетических исследований свидетельствуют о том, что отлаженная работа системы энергоснабжения играет ключевую роль в обеспечении нормальной сердечной деятельности, а селективная делеция компонентов этой системы может спровоцировать развитие СН или ускорить ее прогрессирование.
Однако достоинства генетических исследований одновременно являются и их слабой стороной, поскольку хроническая СН многофакторна и опосредована целым рядом других механизмов, а не только теми, которые контролируются одиночными генами, подвергшимися изучению. Более того, непонятно, каким образом можно деактивировать ген, кодирующий белок с законсервированной в ходе эволюции структурой, которому принадлежит главная роль в процессах энергообмена в миокарде, и не спровоцировать манифестацию СН. Вопрос о том, развиваются ли при «выключении» центральных звеньев энергообеспечения сердечной мышцы какие–то адаптивные реакции, остается открытым.
Перспективные подходы
к лечению СН
Ингибиторы АПФ, диуретики и b–адреноблокаторы могут оказывать опосредованное метаболическое влияние на работу сердца [39,63,109], но прямым действием на энергообмен в миокарде они не обладают. Возникает закономерный вопрос: могут ли процессы энергообмена в сердечной мышце рассматриваться как терапевтическая «мишень» при СН?
Модулирование путей
утилизации субстрата
С точки зрения влияния на энергообмен в миокарде многообещающим подходом выглядит модулирование путей утилизации субстрата. Так, введение в коронарные сосуды пирувата в кратчайшие сроки повышает работоспособность сердца [110], а у собак глюкагоноподобный пептид 1, способствующий утилизации глюкозы, улучшает функции левого желудочка [111]. Наконец, у трансгенных мышей с экспериментальной СН гиперэкспрессия переносчика глюкозы 1 препятствует развитию дисфункции левого желудочка [112].
Непосредственно влиять на пути утилизации субстрата можно с помощью частичных ингибиторов окисления жирных кислот или ингибиторов карнитин–пальмитоил–трансферазы 1. Механизм действия этих ингибиторов сложен [19,21,23], но общим для всех них является то, что они частично подавляют утилизацию жирных кислот, усиливая процессы утилизации глюкозы. Целесообразность применения таких препаратов для лечения СН активно обсуждается; исходы же подобного лечения зависят, по–видимому, от причины, вызвавшей СН, а также от стадии СН. О возможной эффективности частичных ингибиторов окисления жирных кислот свидетельствуют и результаты контрольно–проверочных клинических испытаний. Так, лечение триметазидином, одним из ингибиторов окисления жирных кислот, за 6 месяцев позволяет восстановить функции левого желудочка [113]. Это наблюдение подтверждают и данные 18–месячного исследования, которое проводилось у пациентов с СН, развившейся на фоне инфаркта миокарда [114]. Неподтвержденные результаты небольшого одноцентрового исследования показали, что при СН как ишемического, так и неишемического генеза применение ингибитора окисления жирных кислот пергексилина на протяжении 2 месяцев [115] или ингибитора карнитин–пальмитоил–трансферазы 1 этоксомира на протяжении 3 месяцев улучшает работу левого желудочка. Но эти данные следует интерпретировать с осторожностью, поскольку часть из них была получена без рандомизации, слепого или плацебо–контроля, или же у пациентов с ангинозными болями, что частично объясняет клиническую эффективность торможения процессов окисления жирных кислот. Тем не менее, приведенные сведения подтверждают результаты исследований по изучению эффективности таких ингибиторов при СН у экспериментальных животных [117,118].
Влияние активаторов PRAR на процессы утилизации субстрата в сердечной мышце многопланово. Они могут как напрямую ускорять окисление жирных кислот, так и опосредованно затормозить его за счет снижения уровня липидов в крови. Учитывая, что при гиперэкспрессии PRARa у мышей развивается СН [119], а активаторы PRAR оказывают положительное [120] или отрицательное [121] действие на сердечную мышцу или не влияют на работу сердца вообще [122], можно заключить, что до начала проведения крупномасштабных клинических испытаний лекарственных препаратов на основе PRAR механизмы их влияния на миокард при СН нужно обстоятельно изучить.
Модулирование процессов
окислительного фосфорилирования
Второй подход к проведению метаболической терапии при СН заключается в прямой стимуляции реакций окислительного фосфорилирования. Однако на сегодняшний день эффективных индукторов окислительного фосфорилирования нет. Тем не менее весьма перспективным методом могло бы стать повышение активности PCG–1a, как одного из возможных путей усиления экспрессии ферментов, участвующих в окислительном фосфорилировании [93]. Альтернативой является уменьшение содержания свободных жирных кислот, в результате чего деятельность разобщающих белков будет подавлена, а синтез АТФ, напротив, активируется.
Модулирование метаболизма
макроэргических фосфатов
Еще одна стратегия терапевтических вмешательств в энергообмен – непосредственно повлиять на механизмы запасания макроэргических фосфатов, их доступность и эффективность их утилизации. Усиливая активность белка–переносчика креатина, можно увеличить концентрации креатина и Кф [123]. Хотя резкое усиление активности белка–переносчика креатина нежелательно (поскольку быстрый рост содержания креатина провоцирует подъем уровня свободного АДФ), только последующие исследования помогут установить, насколько целесообразно при СН восстанавливать концентрации креатина и Кф путем умеренной стимуляции переносчика креатина. Наконец, не исключено, что эффективность утилизации АТФ миофибриллами также можно будет повысить с помощью лекарственных препаратов нового поколения – кальций–чувствительных активаторов [124] или активаторов миозина.
Заключение
Метаболическая терапия, «мишенями» для которой служат процессы утилизации субстрата, окислительного фосфорилирования и накопления макроэргических фосфатов, выводит лечение СН на качественно новый этап. Чтобы полностью раскрыть эту концепцию, нужно приложить еще немало усилий. Так, экспериментальные исследования прольют свет на механизмы, лежащие в основе перестройки энергообмена, а также помогут идентифицировать молекулярные «мишени» для терапевтического воздействия. Разработку лекарственных препаратов нового поколения, обладающих модулирующим влиянием, будут вести как научные, так и производственные подразделения. В ходе проведения контрольно–проверочных клинических испытаний для мониторинга ранних изменений энергообмена в миокарде в ответ на метаболическую терапию можно будет использовать такой критерий, как коэффициент соотношения Кф/АТФ, который послужит суррогатным маркером длительного прогностического эффекта. Широкомасштабные клинические испытания позволят доказать или опровергнуть эффективность модуляторов энергообмена в клинике. Не исключено, что все эти достижения уже в ближайшем будущем станут основой для разработки лекарственных препаратов нового поколения, «мишенью» для которых станут процессы энергоснабжения сердечной мышцы. Их внедрение в клиническую практику даст возможность улучшить состояние пациентов и сделать более благоприятным прогноз такого крайне опасного для жизни заболевания, как сердечная недостаточность.
Реферат подготовлен Е.Б. Третьяк
по материалам статьи S. Neubauer
«The failing Heart – an engine out of fuel»
The New England Journal of Medicine
Vol. 356, No.11, March 15, 2007

Литература
1. Heart disease and stroke statistics: 2005 update. Dallas: American Heart Association, 2005.
2. Dayer M, Cowie MR. Heart failure: diagnosis and healthcare burden. Clin Med 2004;4:13–18. [ISI][Medline]
3. McMurray JJ, Pfeffer MA. Heart failure. Lancet 2005;365:1877–1889. [CrossRef][ISI][Medline]
4. The CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med 1987;316:1429–1435. [Abstract]
5. Pfeffer MA, Braunwald E, Moye LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction: results of the Survival and Ventricular Enlargement Trial. N Engl J Med 1992;327:669–677. [Abstract]
6. Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999;341:709–717. [Free Full Text]
7. CIBIS Investigators and Committees. A randomized trial of –blockade in heart failure: the Cardiac Bisoprolol Insufficiency Study (CIBIS). Circulation 1994;90:1765–1773. [Abstract]
8. Packer M, Bristow MR, Cohn JN, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N Engl J Med 1996;334:1349–1355. [Free Full Text]
9. Bristow MR, Saxon LA, Boehmer J, et al. Cardiac–resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med 2004;350:2140–2150. [Free Full Text]
10. Cleland JGF, Daubert J–C, Erdmann E, et al. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med 2005;352:1539–1549. [Free Full Text]
11. Mann DL, Bristow MR. Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation 2005;111:2837–2849. [Erratum, Circulation 2005;112(4):e75.] [Free Full Text]
12. Herrmann G, Decherd GM. The chemical nature of heart failure. Ann Intern Med 1939;12:1233–1244. [ISI]
13. Wollenberger A. On the energy–rich phosphate supply of the failing heart. Am J Physiol 1947;150:733–736. [Free Full Text]
14. Olson RE, Schwartz WB. Myocardial metabolism in congestive heart failure. Medicine (Baltimore) 1951;30:21–41. [ISI][Medline]
15. Olson RE. Myocardial metabolism in congestive heart failure. J Chronic Dis 1959;9:442–464. [CrossRef][Medline]
16. Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res 2004;95:135–145. [Free Full Text]
17. Taegtmeyer H. Metabolism –– the lost child of cardiology. J Am Coll Cardiol 2000;36:1386–1388. [Free Full Text]
18. Taegtmeyer H. Cardiac metabolism as a target for the treatment of heart failure. Circulation 2004;110:894–896. [Free Full Text]
19. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 2005;85:1093–1129. [Free Full Text]
20. Ventura–Clapier R, Garnier A, Veksler V. Energy metabolism in heart failure. J Physiol 2004;555:1–13. [Free Full Text]
21. Morrow DA, Givertz MM. Modulation of myocardial energetics: emerging evidence for a therapeutic target in cardiovascular disease. Circulation 2005;112:3218–3221. [Free Full Text]
22. Lopaschuk GD, Rebeyka IM, Allard MF. Metabolic modulation: a means to mend a broken heart. Circulation 2002;105:140–142. [Free Full Text]
23. Essop MF, Opie LH. Metabolic therapy for heart failure. Eur Heart J 2004;25:1765–1768. [Free Full Text]
24. van Bilsen M, Smeets PJ, Gilde AJ, van der Vusse GJ. Metabolic remodelling of the failing heart: the cardiac burn–out syndrome? Cardiovasc Res 2004;61:218–226. [CrossRef][ISI][Medline]
25. Cohn JN, Tognoni G. A randomized trial of the angiotensin–receptor blocker valsartan in chronic heart failure. N Engl J Med 2001;345:1667–1675. [Free Full Text]
26. Pfeffer MA, Swedberg K, Granger CB, et al. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM–Overall programme. Lancet 2003;362:759–766. [CrossRef][ISI][Medline]
27. Katz AM. Heart failure: pathophysiology, molecular biology and clinical management. Philadelphia: Lippincott Williams & Wilkins, 2000.
28. Bessman SP, Geiger PJ. Transport of energy in muscle: the phosphorylcreatine shuttle. Science 1981;211:448–452. [Free Full Text]
29. Ingwall JS. ATP and the heart. Norwell, MA: Kluwer Academic, 2002.
30. Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the `phosphocreatine circuit' for cellular energy homeostasis. Biochem J 1992;281:21–40. [ISI][Medline]
31. Guimbal C, Kilimann MWA. A Na(+)–dependent creatine transporter in rabbit brain, muscle, heart, and kidney: cDNA cloning and functional expression. J Biol Chem 1993;268:8418–8421. [Free Full Text]
32. Wyss M, Wallimann T. Creatine metabolism and the consequences of creatine depletion in muscle. Mol Cell Biochem 1994;133:51–66. [CrossRef][ISI][Medline]
33. Garlick PB, Radda GK, Seeley PJ. Phosphorus NMR studies on perfused heart. Biochem Biophys Res Commun 1977;74:1256–1262. [CrossRef][ISI][Medline]
34. Ingwall JS. Phosphorus nuclear magnetic resonance spectroscopy of cardiac and skeletal muscles. Am J Physiol 1982;242:H729–H744. [ISI][Medline]
35. Bottomley PA. MR spectroscopy of the human heart: the status and the challenges. Radiology 1994;191:593–612. [Abstract]
36. Neubauer S. Cardiac magnetic resonance spectroscopy. In: Lardo AC, Fayad ZA, Chronos NAF, Fuster V, eds. Cardiovascular magnetic resonance: established and emerging applications. London: Martin Dunitz, 2003:39–60.
37. Neubauer S, Horn M, Cramer M, et al. Myocardial phosphocreatine–to–ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation 1997;96:2190–2196. [Free Full Text]
38. Vitale GD, deKemp RA, Ruddy TD, Williams K, Beanlands RS. Myocardial glucose utilization and optimization of (18)F–FDG PET imaging in patients with non–insulin–dependent diabetes mellitus, coronary artery disease, and left ventricular dysfunction. J Nucl Med 2001;42:1730–1736. [Free Full Text]
39. Wallhaus TR, Taylor M, DeGrado TR, et al. Myocardial free fatty acid and glucose use after carvedilol treatment in patients with congestive heart failure. Circulation 2001;103:2441–2446. [Free Full Text]
40. Davila–Roman VG, Vedala G, Herrero P, et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol 2002;40:271–277. [Free Full Text]
41. Lewandowski ED. Cardiac carbon 13 magnetic resonance spectroscopy: on the horizon or over the rainbow? J Nucl Cardiol 2002;9:419–428. [CrossRef][ISI][Medline]
42. Ning XH, Zhang J, Liu J, et al. Signaling and expression for mitochondrial membrane proteins during left ventricular remodeling and contractile failure after myocardial infarction. J Am Coll Cardiol 2000;36:282–287. [Free Full Text]
43. Gudbjarnason S, Mathes P, Ravens KG. Functional compartmentation of ATP and creatine phosphate in heart muscle. J Mol Cell Cardiol 1970;1:325–339. [CrossRef][Medline]
44. Chandler MP, Kerner J, Huang H, et al. Moderate severity heart failure does not involve a downregulation of myocardial fatty acid oxidation. Am J Physiol Heart Circ Physiol 2004;287:H1538–H1543. [Free Full Text]
45. Osorio JC, Stanley WC, Linke A, et al. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor–alpha in pacing–induced heart failure. Circulation 2002;106:606–612. [Free Full Text]
46. Remondino A, Rosenblatt–Velin N, Montessuit C, et al. Altered expression of proteins of metabolic regulation during remodeling of the left ventricle after myocardial infarction. J Mol Cell Cardiol 2000;32:2025–2034. [CrossRef][ISI][Medline]
47. Nascimben L, Ingwall JS, Lorell BH, et al. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 2004;44:662–667. [Free Full Text]
48. Murakami Y, Zhang Y, Cho YK, et al. Myocardial oxygenation during high work states in hearts with postinfarction remodeling. Circulation 1999;99:942–948. [Free Full Text]
49. Ide T, Tsutsui H, Hayashidani S, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 2001;88:529–535. [Free Full Text]
50. Marin–Garcia J, Goldenthal MJ, Moe GW. Abnormal cardiac and skeletal muscle mitochondrial function in pacing–induced cardiac failure. Cardiovasc Res 2001;52:103–110. [CrossRef][ISI][Medline]
51. Pool PE, Spann JF Jr, Buccino RA, Sonnenblick EH, Braunwald E. Myocardial high energy phosphate stores in cardiac hypertrophy and heart failure. Circ Res 1967;21:365–373. [ISI][Medline]
52. Bittner V, Reeves RC, Digerness SB, Caulfield JB, Pohost GM. 31P NMR spectroscopy in chronic adriamycin cardiotoxicity. Magn Reson Med 1991;17:69–81. [CrossRef][ISI][Medline]
53. Nascimben L, Friedrich J, Liao R, Pauletto P, Pessina AC, Ingwall JS. Enalapril treatment increases cardiac performance and energy reserve via the creatine kinase reaction in myocardium of Syrian myopathic hamsters with advanced heart failure. Circulation 1995;91:1824–1833. [Free Full Text]
54. Neubauer S, Horn M, Naumann A, et al. Impairment of energy metabolism in intact residual myocardium of rat hearts with chronic myocardial infarction. J Clin Invest 1995;95:1092–1100. [ISI][Medline]
55. Zhang J, Merkle H, Hendrich K, et al. Bioenergetic abnormalities associated with severe left ventricular hypertrophy. J Clin Invest 1993;92:993–1003. [ISI][Medline]
56. Zhang J, Wilke N, Wang Y, et al. Functional and bioenergetic consequences of postinfarction left ventricular remodeling in a new porcine model: MRI and 31 P–MRS study. Circulation 1996;94:1089–1100. [Free Full Text]
57. Zhang J, Toher C, Erhard M, et al. Relationships between myocardial bioenergetic and left ventricular function in hearts with volume–overload hypertrophy. Circulation 1997;96:334–343. [Free Full Text]
58. Kalsi KK, Smolenski RT, Pritchard RD, Khaghani A, Seymour AM, Yacoub MH. Energetics and function of the failing human heart with dilated or hypertrophic cardiomyopathy. Eur J Clin Invest 1999;29:469–477. [CrossRef][ISI][Medline]
59. Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation 2001;104:2923–2931. [Free Full Text]
60. Taylor M, Wallhaus TR, Degrado TR, et al. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F]fluoro–6–thia–heptadecanoic acid and [18F]FDG in patients with congestive heart failure. J Nucl Med 2001;42:55–62. [Free Full Text]
61. Quigley AF, Kapsa RM, Esmore D, Hale G, Byrne E. Mitochondrial respiratory chain activity in idiopathic dilated cardiomyopathy. J Card Fail 2000;6:47–55. [CrossRef][ISI][Medline]
62. Hardy CJ, Weiss RG, Bottomley PA, Gerstenblith G. Altered myocardial high–energy phosphate metabolites in patients with dilated cardiomyopathy. Am Heart J 1991;122:795–801. [CrossRef][ISI][Medline]
63. Neubauer S, Krahe T, Schindler R, et al. 31P magnetic resonance spectroscopy in dilated cardiomyopathy and coronary artery disease: altered cardiac high–energy phosphate metabolism in heart failure. Circulation 1992;86:1810–1818. [Free Full Text]
64. Regitz V, Fleck E. Myocardial adenine nucleotide concentrations and myocardial norepinephrine content in patients with heart failure secondary to idiopathic dilated or ischemic cardiomyopathy. Am J Cardiol 1992;69:1574–1580. [CrossRef][ISI][Medline]
65. Starling RC, Hammer DF, Altschuld RA. Human myocardial ATP content and in vivo contractile function. Mol Cell Biochem 1998;180:171–177. [CrossRef][ISI][Medline]
66. Beer M, Seyfarth T, Sandstede J, et al. Absolute concentrations of high–energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P–SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol 2002;40:1267–1274. [Free Full Text]
67. Nascimben L, Ingwall JS, Pauletto P, et al. Creatine kinase system in failing and nonfailing human myocardium. Circulation 1996;94:1894–1901. [Free Full Text]
68. Conway MA, Allis J, Ouwerkerk R, Niioka T, Rajagopalan B, Radda GK. Detection of low phosphocreatine to ATP ratio in failing hypertrophied human myocardium by 31P magnetic resonance spectroscopy. Lancet 1991;338:973–976. [CrossRef][ISI][Medline]
69. Casademont J, Miro O. Electron transport chain defects in heart failure. Heart Fail Rev 2002;7:131–139. [CrossRef][Medline]
70. Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K. Uncoupling proteins in human heart. Lancet 2004;364:1786–1788. [CrossRef][ISI][Medline]
71. Shen W, Asai K, Uechi M, et al. Progressive loss of myocardial ATP due to a loss of total purines during the development of heart failure in dogs: a compensatory role for the parallel loss of creatine. Circulation 1999;100:2113–2118. [Free Full Text]
72. Ten Hove M, Chan S, Lygate C, et al. Mechanisms of creatine depletion in chronically failing rat heart. J Mol Cell Cardiol 2005;38:309–313. [CrossRef][Medline]
73. Neubauer S, Remkes H, Spindler M, et al. Downregulation of the Na(+)–creatine cotransporter in failing human myocardium and in experimental heart failure. Circulation 1999;100:1847–1850. [Free Full Text]
74. Vatner DE, Ingwall JS. Effects of moderate pressure overload cardiac hypertrophy on the distribution of creatine kinase isozymes. Proc Soc Exp Biol Med 1984;175:5–9. [Abstract]
75. Ingwall JS, Kramer MF, Fifer MA, et al. The creatine kinase system in normal and diseased human myocardium. N Engl J Med 1985;313:1050–1054. [Abstract]
76. Laser A, Ingwall JS, Tian R, et al. Regional biochemical remodeling in non–infarcted tissue of rat heart post–myocardial infarction. J Mol Cell Cardiol 1996;28:1531–1538. [CrossRef][ISI][Medline]
77. Khuchua ZA, Ventura–Clapier R, Kuznetsov AV, Grishin MN, Saks VA. Alterations in the creatine kinase system in the myocardium of cardiomyopathic hamsters. Biochem Biophys Res Commun 1989;165:748–757. [CrossRef][ISI][Medline]
78. De Sousa E, Veksler V, Minajeva A, et al. Subcellular creatine kinase alterations: implications in heart failure. Circ Res 1999;85:68–76. [Free Full Text]
79. Ye Y, Gong G, Ochiai K, Liu J, Zhang J. High–energy phosphate metabolism and creatine kinase in failing hearts: a new porcine model. Circulation 2001;103:1570–1576. [Free Full Text]
80. Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci U S A 2005;102:808–813. [Free Full Text]
81. Liao R, Nascimben L, Friedrich J, Gwathmey JK, Ingwall JS. Decreased energy reserve in an animal model of dilated cardiomyopathy: relationship to contractile performance. Circ Res 1996;78:893–902. [Free Full Text]
82. Liu J, Wang C, Murakami Y, et al. Mitochondrial ATPase and high–energy phosphates in failing hearts. Am J Physiol Heart Circ Physiol 2001;281:H1319–H1326. [Free Full Text]
83. Neubauer S, Horn M, Pabst T, et al. Contributions of 31P–magnetic resonance spectroscopy to the understanding of dilated heart muscle disease. Eur Heart J 1995;16:Suppl O:115–118. [ISI][Medline]
84. Lamb HJ, Beyerbacht HP, van der Laarse A, et al. Diastolic dysfunction in hypertensive heart disease is associated with altered myocardial metabolism. Circulation 1999;99:2261–2267. [Free Full Text]
85. Ashrafian H, Redwood C, Blair E, Watkins H. Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet 2003;19:263–268. [CrossRef][ISI][Medline]
86. Crilley JG, Boehm EA, Blair E, et al. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol 2003;41:1776–1782. [Free Full Text]
87. Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res 2004;95:568–578. [Free Full Text]
88. Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of peroxisome proliferator–activated receptor–alpha during cardiac hypertrophic growth. J Clin Invest 2000;105:1723–1730. [Free Full Text]
89. Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 1996;94:2837–2842. [Free Full Text]
90. Karbowska J, Kochan Z, Smolenski RT. Peroxisome proliferator–activated receptor alpha is downregulated in the failing human heart. Cell Mol Biol Lett 2003;8:49–53. [ISI][Medline]
91. Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura–Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 2003;551:491–501. [Free Full Text]
92. Finck BN, Kelly DP. PGC–1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest 2006;116:615–622. [Free Full Text]
93. Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR–gamma coactivator 1alpha. Proc Natl Acad Sci U S A 2006;103:10086–10091. [Free Full Text]
94. Exil VJ, Roberts RL, Sims H, et al. Very–long–chain acyl–coenzyme A dehydrogenase deficiency in mice. Circ Res 2003;93:448–455. [Free Full Text]
95. Kurtz DM, Rinaldo P, Rhead WJ, et al. Targeted disruption of mouse long–chain acyl–CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc Natl Acad Sci U S A 1998;95:15592–15597. [Free Full Text]
96. Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R. Decreased contractile and metabolic reserve in peroxisome proliferator–activated receptor–alpha–null hearts can be rescued by increasing glucose transport and utilization. Circulation 2005;112:2339–2346. [Free Full Text]
97. Abel ED, Kaulbach HC, Tian R, et al. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest 1999;104:1703–1714. [Free Full Text]
98. Arany Z, He H, Lin J, et al. Transcriptional coactivator PGC–1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab 2005;1:259–271. [CrossRef][ISI][Medline]
99. Hansson A, Hance N, Dufour E, et al. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain–deficient mouse hearts. Proc Natl Acad Sci U S A 2004;101:3136–3141. [Free Full Text]
100. Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet 1997;16:226–234. [CrossRef][ISI][Medline]
101. Nahrendorf M, Spindler M, Hu K, et al. Creatine kinase knockout mice show left ventricular hypertrophy and dilatation, but unaltered remodeling post–myocardial infarction. Cardiovasc Res 2005;65:419–427. [CrossRef][ISI][Medline]
102. Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A, Ventura–Clapier R. Energetic crosstalk between organelles: architectural integration of energy production and utilization. Circ Res 2001;89:153–159. [Free Full Text]
103. ten Hove M, Lygate CA, Fischer A, et al. Reduced inotropic reserve and increased susceptibility to cardiac ischemia/reperfusion injury in phosphocreatine–deficient guanidinoacetate–N–methyltransferase–knockout mice. Circulation 2005;111:2477–2485. [Free Full Text]
104. Chapoy PR, Angelini C, Brown WJ, Stiff JE, Shug AL, Cederbaum SD. Systemic carnitine deficiency –– a treatable inherited lipid–storage disease presenting as Reye's syndrome. N Engl J Med 1980;303:1389–1394. [Abstract]
105. Matalon R, Michaels K, Kaul R, et al. Malonic aciduria and cardiomyopathy. J Inherit Metab Dis 1993;16:571–573. [CrossRef][ISI][Medline]
106. Hug G, Bove KE, Soukup S. Lethal neonatal multiorgan deficiency of carnitine palmitoyltransferase II. N Engl J Med 1991;325:1862–1864. [ISI][Medline]
107. Guertl B, Noehammer C, Hoefler G. Metabolic cardiomyopathies. Int J Exp Pathol 2000;81:349–372. [CrossRef][ISI][Medline]
108. Russell LK, Finck BN, Kelly DP. Mouse models of mitochondrial dysfunction and heart failure. J Mol Cell Cardiol 2005;38:81–91. [CrossRef][ISI][Medline]
109. Hugel S, Horn M, de Groot M, et al. Effects of ACE inhibition and beta–receptor blockade on energy metabolism in rats postmyocardial infarction. Am J Physiol 1999;277:H2167–H2175. [ISI][Medline]
110. Hermann HP, Pieske B, Schwarzmuller E, Keul J, Just H, Hasenfuss G. Haemodynamic effects of intracoronary pyruvate in patients with congestive heart failure: an open study. Lancet 1999;353:1321–1323. [CrossRef][ISI][Medline]
111. Nikolaidis LA, Elahi D, Hentosz T, et al. Recombinant glucagon–like peptide–1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing–induced dilated cardiomyopathy. Circulation 2004;110:955–961. [Free Full Text]
112. Liao R, Jain M, Cui L, et al. Cardiac–specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 2002;106:2125–2131. [Free Full Text]
113. Vitale C, Wajngaten M, Sposato B, et al. Trimetazidine improves left ventricular function and quality of life in elderly patients with coronary artery disease. Eur Heart J 2004;25:1814–1821. [Free Full Text]
114. Di Napoli P, Taccardi AA, Barsotti A. Long term cardioprotective action of trimetazidine and potential effect on the inflammatory process in patients with ischaemic dilated cardiomyopathy. Heart 2005;91:161–165. [Free Full Text]
115. Lee L, Campbell R, Scheuermann–Freestone M, et al. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short–term use of a novel treatment. Circulation 2005;112:3280–3288. [Free Full Text]
116. Schmidt–Schweda S, Holubarsch C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci (Lond) 2000;99:27–35. [Medline]
117. Lionetti V, Linke A, Chandler MP, et al. Carnitine palmitoyl transferase–I inhibition prevents ventricular remodeling and delays decompensation in pacing–induced heart failure. Cardiovasc Res 2005;66:454–461. [CrossRef][ISI][Medline]
118. Chandler MP, Stanley WC, Morita H, et al. Short–term treatment with ranolazine improves mechanical efficiency in dogs with chronic heart failure. Circ Res 2002;91:278–280. [Free Full Text]
119. Finck BN, Han X, Courtois M, et al. A critical role for PPARalpha–mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A 2003;100:1226–1231. [Free Full Text]
120. Shiomi T, Tsutsui H, Hayashidani S, et al. Pioglitazone, a peroxisome proliferator–activated receptor–gamma agonist, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 2002;106:3126–3132. [Free Full Text]
121. Lygate CA, Hulbert K, Monfared M, Cole MA, Clarke K, Neubauer S. The PPARgamma–activator rosiglitazone does not alter remodeling but increases mortality in rats post–myocardial infarction. Cardiovasc Res 2003;58:632–637. [CrossRef][ISI][Medline]
122. Frantz S, Hu K, Widder J, et al. Peroxisome proliferator activated–receptor agonism and left ventricular remodeling in mice with chronic myocardial infarction. Br J Pharmacol 2004;141:9–14. [CrossRef][ISI][Medline]
123. Wallis J, Lygate CA, Fischer A, et al. Supranormal myocardial creatine and phosphocreatine concentrations lead to cardiac hypertrophy and heart failure: insights from creatine transporter–overexpressing transgenic mice. Circulation 2005;112:3131–3139. [Free Full Text]
124. Ng TM. Levosimendan, a new calcium–sensitizing inotrope for heart failure. Pharmacotherapy 2004;24:1366–1384. [CrossRef][ISI][Medline]



