




Методы
Поиск по литературным базам научных данных Medline и EMBASE проводился в августе 2008 г. без ограничений по дате публикаций. В качестве параметров поиска были заданы полнотекстовые статьи на английском языке, содержащие как ключевые слова (амлодипин, биодоступность, стабильность, токсичность, генерические препараты, терапевтическая эквивалентность), так и текстонезависимые термины (амлодипина безилат, амлодипина малеат, эквивалентность, руководства, соли). Кроме того, проанализирована и библиография по списку литературы. Результаты поиска по критериям «биоэквивалентность» и «терапевтическая эквивалентность» не рассматривалась в качестве основы для составлении систематического обзора. Тем не менее приводимый ниже обзор все же можно считать систематическим, поскольку он включает все доступные научные данные по амлодипина малеату (с учетом заданных ограничений). Также представлена информация с ведущих европейских и американских веб–сайтов, посвященных этой проблеме.
Терминология понятий
«эквивалентность» и «сходство»
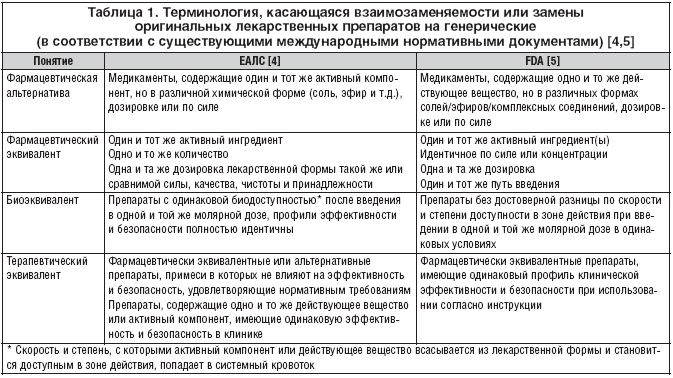
Несмотря на некоторые различия в формулировке, эксперты как Европейского агентства лекарственных средств (ЕАЛС), так и Управления по контролю за продуктами и лекарствами США (FDA) определяют понятия «фармацевтическая альтернатива» и «фармацевтическая эквивалентность» сходным образом (табл. 1). Генерические препараты – альтернативные или эквивалентные – имеют такой же состав активных ингредиентов, как и оригинальное лекарственное средство. Однако они могут отличаться от него по форме, размеру, цвету, конфигурации насечек (рисок) на плоской поверхности, механизму высвобождения (немедленное, модифицированное и т.д.), вспомогательным веществам (красители, отдушки, консерванты, связующие вещества, наполнители, смазочные материалы, дезинтегрирующие агенты и т.д.), по способу производства, сроку годности, виду упаковки и, с некоторыми ограничениями, маркировке [3]. Допускается различный состав наполнителей, которые, как предполагается, должны быть инертными, однако генерик должен иметь такое же соотношение действующих и вспомогательных компонентов, как и оригинальный препарат [1].
Согласно определению под биоэквивалентностью подразумевают отсутствие между препаратами достоверной разницы по скорости и степени всасывания (т.е. биодоступности) при их использовании в одной и той же молярной дозе (табл. 1) [4,5]. Биоэквивалентные препараты считаются «существенно однородными», что признано FDA [6]. Они «имеют одинаковый качественный и количественный состав (имеется в виду содержание активных действующих веществ), лекарственную форму и биоэквивалентны в той степени, пока научными исследованиями не доказано, что препарат с медицинальной прописью отличается от оригинального по своей эффективности и безопасности» [4]. Как это ни парадоксально, но в руководстве ЕАЛС встречаются две трактовки терапевтической эквивалентности (табл. 1) [4]: фармацевтически эквивалентные лекарственные препараты считаются терапевтически эквивалентными при наличии доказанной биоэквивалентности, но в случае фармацевтически альтернативных лекарственных средств могут потребоваться дополнительные (до)клинические данные, которые позволяли бы говорить о их терапевтической эквивалентности [7].
Все эти термины отражены в законодательных требованиях к использованию генерических лекарственных средств. Если речь идет о фармацевтически эквивалентных лекарствах, то для них применима сокращенная процедура регистрации новых препаратов (ANDA) [8]. Подавая заявку на ANDA, спонсор должен представить доказательства биоэквивалентности фармацевтически эквивалентного генерика и запатентованного препарата (рис. 1), которые, согласно определению, терапевтически эквивалентны [8]. В отличие от заявки на новый препарат (NDA), при подаче которой предъявляются высокие требования к качеству, в случае ANDA необязательно представлять данные о клинической безопасности и эффективности (рис. 1) [8,9].
Оценка и критерии
биоэквивалентности
Несмотря на то, что в разных странах используются различные методы оценки биоэквивалентности [10], в руководствах Всемирной Организации Здравоохранения (ВОЗ) приводится общая рекомендация – включать в перекрестное исследование, как минимум, 12 клинически здоровых взрослых добровольцев в возрасте 18–55 лет с нормальной массой тела [2]. На практике биоэквивалентность изучают в рандомизированных перекрестных двухэтапных исследованиях в группах из 18–24 клинически здоровых и относительно молодых добровольцев. Обычно однократную дозу генерического или оригинального препарата принимают, соблюдая стандартные условия (с учетом характера питания, количества потребляемой жидкости, уровня физической нагрузки и времени приема препарата). Чтобы минимизировать вариабельность между субъектами, формируют стандартные выборки и используют стандартизированные протоколы, в результате чего любые возникающие отклонения, выходящие за рамки статистически допустимых, можно отнести за счет разницы прописей, а не индивидуальных особенностей испытуемых [11]. Более того, полученные данные позволяют предполагать, что оценка системного высвобождения активного компонента оказывается более чувствительной, если проводятся исследования с однократной дозой, а не многократными [11]. Поскольку одновременный прием пищи и пероральных лекарственных препаратов может повлиять на биоэквивалентность [6,11], рекомендуется (в случае лекарственных средств с пролонгированным действием) [5] или даже требуется (в случае взаимодействия лекарственного средства и компонентов пищи) [4] осуществлять дополнительное тестирование ингредиентов, входящих в состав стандартизированного набора продуктов питания.
Фармакокинетические эффекты лекарственных препаратов оцениваются и статистически анализируются по таким параметрам, как площадь под кривой концентрации в плазме крови в зависимости от времени (AUC) и максимальная концентрация в плазме крови (Cmax) [4,5]. Эти показатели позволяют наиболее точно определить степень и скорость всасывания препарата (т.е. биодоступность) и его экспозицию [4,5], конечный период полураспада (t1/2) [4, 5], константу скорости элиминации (λZ) [5] и – в специфических обстоятельствах – скорость экскреции с мочой (АС) [4]. О биоэквивалентности можно говорить в том случае, если 90% доверительный интервал (ДИ) AUC и Cmax для соотношения генерик/оригинальный препарат находится в границах значений от 0,80 до 1,25 [4,5]. Поскольку для сравнения данные логарифмируются, возникает асимметрия, получившая название «правило –20%/+25%» [13]. Однако для лекарственных препаратов с критической дозой, которые имеют узкий терапевтический индекс (т.е. небольшую разницу между минимальной эффективной концентрацией и минимальной токсической концентрацией) – иммунодепрессантов, антиэпилептических средств, сердечных гликозидов (дигоксин), антикоагулянтов (варфарин) [12] – пределы этих величин сокращаются [4]. Это связано с тем, что даже относительно небольшие колебания системного уровня таких лекарственных веществ могут спровоцировать заметное изменение фармакодинамики, а именно – их эффективности или частоты встречаемости побочного действия [11]. В случае препаратов с высокой вариабельностью внутри субъектов (>30%) и небольшой токсичностью при достижении Cmax ЕАЛС (но не FDA) допускает расширение границ 90% ДИ Cmax до 0,75–1,33 [14,15]. Необходимость оценивать tmax для установления биоэквивалентности определяется регулирующими законами. Отчасти это объясняется отсутствием единых статистических методов анализа tmax – величины, которая (в отличие от непрерывных переменных AUC и Cmax) является дискретной и зависит от плана выборочного контроля, оговоренного протоколом. Таким образом, в отличие от FDA ЕАЛС требует определять tmax лишь при наличии клинически значимых указаний на быстрое высвобождение/начало действия или при возникновении признаков побочного действия [4].
Разногласия по вопросу
взаимозаменяемости
ЕАЛС не дает никаких четких рекомендаций относительно взаимозаменяемости биоэквивалентных лекарств. По сведениям FDA, в США около 20% генериков не являются биоэквивалентами зарегистрированных торговых брендов, а следовательно, эти препараты не могут считаться взаимозаменяемыми [11]. Но как это ни парадоксально, эксперты FDA указывают на отсутствие документально подтвержденных данных о том, что конкретный генерик не может заменить соответствующий запатентованный оригинальный препарат [16]. Таким образом, врачам не приходится беспокоиться, если пациент отказывается от оригинального лекарственного средства и переходит на прием генерического (или меняет один генерик на другой).
Тем не менее, с учетом расхождений в терминологии биоэквивалентности и подходах к ее оценке, а также в критериях терапевтической эквивалентности (что ставит вопрос о терапевтической эффективности препарата) [10], представляется целесообразным рассмотреть различные аспекты взаимозаменяемости.
Доказательность исследований
по биоэквивалентности
Как правило, результаты исследований по биоэквивалентности используются организациями здравоохранения для разработки регулирующих положений, однако публикуются крайне редко [17]. Обычно эти данные находятся в свободном доступе на соответствующих веб–сайтах или могут быть получены благодаря Закону о свободе информации (если это данные американских исследований), но частично ограниченный доступ все же исключает возможность их легкого анализа и проверки широкой научной общественностью.
Выводы о биоэквивалентности тех или иных лекарств базируются в основном на результатах относительно небольших испытаний с приемом фиксированных доз препаратов, к которым привлекаются клинически здоровые добровольцы. Следовательно, в ходе таких исследований не достигается равновесная концентрация лекарственных средств. Но в случае большинства хронических заболеваний для достижения терапевтического эффекта необходимо не просто достигать такой концентрации препарата, но и длительно поддерживать ее [11]. Если пациент находится на поддерживающей терапии, то уровень лекарства в его крови обычно выше, чем после приема однократной дозы (порой в несколько раз) [18]. Таким образом, в ходе проведения исследований с привлечением клинически здоровых добровольцев получаемые данные не отражают реальных ситуаций, наблюдаемых в клинической практике. Это может вызвать определенные сложности, поскольку не исключено, что в процессе поддерживающей терапии фармакокинетика препарата меняется под действием гипотетически инертных вспомогательных веществ (наполнителей) и примесей и/или в результате накопления активных метаболитов [11]. Кроме того, характеристики однородной группы клинически здоровых добровольцев и выборки больных, скорее всего, будут различаться (в последнем случае это лица более пожилого возраста с разными сопутствующими заболеваниями, принимающие разнообразные лекарственные средства от гипертонии и/или ишемической болезни сердца), и поэтому экстраполировать данные нельзя [11]. Фармакокинетика препарата также может меняться на фоне физиологических процессов старения, в результате взаимодействия с используемыми одновременно лекарствами и/или из–за наличия сопутствующих заболеваний. Следовательно, нельзя сравнивать воздействие лекарственного вещества на здорового человека и эффекты того же самого препарата в повседневной клинической практике [11]. Характерные примеры – гидрохлорид прокаина, степень всасывания которого у клинически здоровых лиц и больных с острым инфарктом миокарда статистически достоверно отличается [19], и генерик верапамила, который биоэквивалентен оригинальному продукту только у молодых и клинически здоровых людей, но не у пожилых пациентов [20].
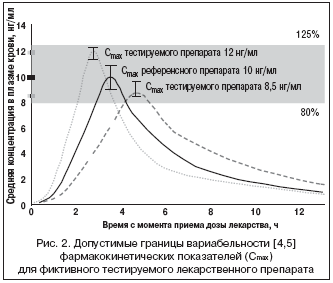
Далее, подвергаются критике и значения эквивалентности в пределах от 0,8 до 1,25, ибо теоретически скорость и/или степень всасывания сравниваемых препаратов в действительности может отличаться на 20% (рис. 2) [1]. Для лекарств с зарегистрированным торговым названием стандарты гораздо строже (5%) [21], а для препаратов с узким терапевтическим индексом требования упрощаются [4,5]. Небольшие отличия в биодоступности приобретают значение в тех случаях, когда лекарственное средство плохо растворяется в воде, имеет нелинейную кинетику и/или модифицированный профиль высвобождения [11,13].
Существует и более фундаментальная проблема, связанная с тем, что истинно биоэквивалентные препараты оказывают на конкретного больного одинаковый эффект (т.е. являются терапевтически эквивалентными) [13]. Но на практике определить это не представляется возможным, поскольку в исследованиях по изучению биоэквивалентности анализируются усредненные значения для генериков и оригинальных продуктов, отражающие среднюю эквивалентность по показателю биодоступности [13]. Однако этого недостаточно, чтобы судить о том, насколько взаимозаменяемы препараты [22]. Более достоверными можно считать результаты, которые получают, используя другие подходы – популяционный или индивидуальный [11]: они позволяют оценить не только среднюю биоэквивалентность, но и равенство в распределении биодоступности внутри субъектов и между ними [11]. Тем не менее регулятивные органы не санкционируют применение альтернативных подходов, их внедрение допускается только в особых ситуациях [23].
Регулятивные нормы понятия
«терапевтическая эквивалентность»
Говоря о терапевтической эквивалентности, которую устанавливают по биоэквивалентности, необходимо отметить, что идентичность наполнителей не является строго обязательным условием. Однако состав последних играет важную роль в обеспечении стабильности и сохранении внешнего вида продукта [1], а следовательно, различия в содержании наполнителей могут обусловливать несоответствие терапевтического действия и профиля безопасности/толерантности [11,24]. Кроме того, срок хранения таблетированных форм лекарственных средств зависит от особенностей процесса их производства (уровень компрессионного давления, использование вращающихся или иных машин и т.д.) [1]. В процессе проведения большинства исследований по биоэквивалентности эти аспекты вряд ли учитываются.
Хорошо известно, что с точки зрения исходов лечения далеко не все препараты одного и того же терапевтического класса являются взаимозаменяемыми [25], и это может быть обусловлено целым рядом факторов. Генерические и оригинальные продукты в данном случае не исключение. Так, все гипотензивные лекарства регистрируются на основании того, что снижают артериальное давление (АД). Предполагается, что при уменьшении АД на какую–то конкретную величину эти препараты в одинаковой мере повлияют на фиксированные конечные точки, сокращая риск (не)фатального инсульта, ИМ или сердечной недостаточности. Но если генерик содержит, например, другую соль активного компонента, то это допущение может оказаться неверным. Поэтому для установления взаимозаменяемости лекарственных веществ более целесообразно их непосредственное сравнение в течение продолжительного времени, рассматривая в качестве первичной конечной точки частоту встречаемости тех или иных клинических явлений [25]. Хотя это не относится абсолютно ко всем генерикам, следует подчеркнуть, что для них гораздо больше подходят не современные рекомендации, а регулятивные нормы по биологически сходным препаратам (т.е. биологическим препаратам, генерикам и медицинским продуктам, произведенным с помощью биотехнологий). Согласно руководствам ЕАЛС по этим веществам, перед регистрацией на фармацевтическом рынке нужно провести их (до)клинические испытания [26].
Соль активного компонента
как ключевой фактор
Альтернативные соли запатентованных лекарственных средств рассматриваются ЕАЛС и FDA, как новые химические соединения [7]. И все же процедура регистрации таких препаратов существенно упрощается благодаря предшествующему (клиническому) опыту использования других солей [7]. Если достоверно установлено, что фармакокинетика, фармакодинамика и/или токсичность действующего вещества препарата, в состав которого входит другой вид соли, не меняется (а эти факторы способны повлиять на эффективность и/или безопасность лекарства), то применима сокращенная процедура подача заявки по форме 505b(2), или гибридная NDA [7].
Примерно половина действующих веществ препаратов для терапевтического использования – это соли (а не свободные кислоты или основания) [7]. Синтез альтернативных видов солей лекарственных средств является методом оптимизации их физико–химических свойств – таких как растворимость, гигроскопичность, (термо)стабильность, растворяемость, текучесть, механизм деградации – без изменения структуры [7]. Но эти же свойства определяют, в какой степени лекарство задерживается в организме, а следовательно, форма соли может повлиять на его биологические характеристики (т.е. фармакокинетику и фармакодинамику), клиническую эффективность [27]. В настоящее время не существует никаких надежных способов, которые позволили бы с точностью прогнозировать, как скажется изменение вида соли на состоянии активной субстанции [27].
Чтобы иметь возможность подать заявку на ANDA, содержащую информацию по биоэквивалентности [7], и получить официальную регистрацию на рынке фармацевтических товаров до истечения срока действия патента оригинального продукта [28], при производстве генериков фармацевтические компании зачастую используют другие виды солей. Такие генерики не следует автоматически считать фармацевтическим эквивалентом оригинального препарата, скорее, их нужно рассматривать, как фармацетическую альтернативу, т.е. химическое производное действующего вещества. Из этого логически вытекает, что о терапевтической эквивалентности таких генериков нельзя судить только на основании данных по биоэквивалентности, а для их широкого внедрения в практическую деятельность необходимы дополнительные доклинические и клинические испытания [7].
Влияние на всасываемость,
переносимость и безопасность
Еще один фактор, влияющий на биодоступность лекарства благодаря изменению его растворимости [39] – это полиморфизм твердого тела. Он определяется, как способность вещества поддерживать строго упорядоченную конформацию и/или порядок расположения молекул при нахождении в кристаллическом состоянии.
Соли различаются по своей растворимости в воде и скорости растворения. Эти характеристики детерминируют степень всасывания препарата in vivo, а следовательно, его фармакокинетику и биологические свойства [27]. Это лишний раз свидетельствует о необходимости проведения исследований по биоэквивалентности, хотя вопросы переносимости и безопасности лекарств в них рассматриваются не всегда. Например, конъюгированные катионы или анионы солеобразующих агентов могут реагировать с солями, вызывая тем самым токсический эффект [7]. Эти данные были получены в ходе доклинических испытаний правадолина малеата, нефротоксичность которого, как показано, обусловлена образованием малеиновой кислоты [30]. Изменение вида соли может иметь и иные непредсказуемые последствия. Так, нарушение работы желудочно–кишечного тракта на фоне использования некоторых солей алпренолола, выявляемые у подопытных животных в эзофагальном тесте, связывают с повышением его растворимости [31]. Наконец, могут нарушаться и лекарственные взаимодействия: установлено, что обезболивающее вещество пропоксифена гидрохлорид дестабилизирует ацетилсалициловую кислоту [27].
Влияние на стабильность
и оптимальную пропись
Гигроскопичность и гидрофобность соли отчасти определяет стабильность активной субстанции лекарства, особенно если оно легко подвергается гидролизу [27]. В случае низкой точки плавления соли происходит пластическая деформация препарата с последующим затвердеванием или агрегацией действующего вещества [7]. В результате дозировка лекарственного средства перестает быть универсальной, ухудшаются и другие характеристики твердой лекарственной формы, что отрицательно влияет на процесс промышленного производства [7].
Биологически активные примеси
Химические примеси, появляющиеся в процессе синтеза того или иного лекарства или из–за его нестабильности, могут спровоцировать токсические явления при его употреблении [7]. Поэтому содержание примесей не должно превышать допустимые нормы, прописанные в нормативных документах Международной конференции по согласованию технических требований к регистрации медикаментов, применяемых у человека [32,33].
Нестабильность препарата вследствие изменения формы соли можно продемонстрировать на примере амлодипина малеата (рис. 3) [11]. В отличие от безилата (рис. 3) малеат подвергается деградации, в результате которой образуются химические примеси [7]. Одна из таких реакций – присоединение первичной аминной группы амлодипина к ненасыщенной малеиновой кислоте [7]. Эта побочная реакция имеет место как на этапе синтеза соли действующего вещества лекарства, так и в процессе производства и хранения готовой продукции [34]. При изучении стабильности экспериментальных фармакологических прописей было установлено, что содержание примесей может достигать 2% [34]. Непонятно, имеет ли это какую–то клиническую значимость, однако точно известно, что биологическая активность этих примесей не соответствует характеристикам амлодипина [34]. Результаты лигандного и ферментного анализа очищенных (>99%) продуктов деградации (100 нМ) свидетельствуют о широком спектре опосредованных ими молекулярных и тканевых эффектов, в том числе ухудшение сократимости изолированной сердечной мышцы [34].
Далее, при проведении высокоэффективной жидкостной хроматографии в составе амлодипина малеата были обнаружены 6 видов примесей в количестве от 0,43 до 1,42% [35]. В таблетках амлодипина малеата (но не безилата) были выявлены два основных продукта деградации, что лишний раз подтверждает гипотезу о различном профиле стабильности этих лекарственных соединений [36]. Таким образом, присущая амлодипина малеату нестабильность, обусловливающая появление в готовой лекарственной форме примесей (т.е. биологически активных продуктов деградации), не позволяет говорить об эквивалентности малеиновой и безилатной солей амлодипина [7].
Примеси и продукты деградации как результат изменения формы соли активного компонента потенциально могут оказывать и генотоксичный эффект. Недавно Комитетом по медицинским препаратам для клинического использования ЕАЛС было выпущено отдельное руководство по генотоксичным примесям [31]. В нем представлены общая схема и практические рекомендации того, как нивелировать действие генотоксичных примесей, которые содержатся к лекарственных средствах, синтезированных на основе новых действующих веществ. В США, Канаде и Японии таких руководств нет, и пути решения проблемы пока не найдены.
Амлодипина безилат и амлодипина малеат: краткая справка
Клинические данные по амлодипина безилату
Механизм действия амлодипина – дигидропиридинового антагониста кальция – заключается в расслаблении гладкомышечных клеток сосудистой стенки и уменьшении периферического сосудистого сопротивления, следствием чего является снижение системного АД [38,39]. Благодаря способности вызывать дилатацию периферических и коронарных сосудов [38] он купирует приступ стенокардии, что также отчасти связано со снижением потребности миокарда в кислороде и падением тонуса коронарных сосудов (т.е. снятием их спазма). Все это в совокупности обусловливает восстановление коронарного кровотока [38,39].
В 1992 г. компания Pfizer выпустила амлодипина безилат в таблетированной форме для приема 1 раз в сутки (дозировка 2,5–5–10 мг), зарегистрировав его под торговыми названиями Норваск (США и большинство стран Европы), Истин (Великобритания, Ирландия) и Амлор (Бельгия, Франция). Показаниями к назначению препаратов амлодипина служат артериальная гипертония, хроническая стабильная стенокардия и вазоспастическая стенокардия (Принцметала или вариантная) [39–42].
Клинический профиль амлодипина активно изучался как на этапе его разработки, так и после регистрации. Особое внимание уделялось его фармакологическим свойствам, а также безопасности и эффективности на отдаленных сроках (с анализом фиксированных конечных точек). Практически все данные по безопасности и эффективности амлодипина касаются его безилатной соли. Результаты недавно завершенного мета–анализа свидетельствуют о том, что в качестве средства профилактики инсульта, как одного из сердечно–сосудистых заболеваний, амлодипина безилат эффективнее других гипотензивных препаратов и плацебо (соотношение рисков 0,81 при p<0,0001 и 0,63 при p=0,06, соответственно) [43]. Лечение амлодипина безилатом также значительно уменьшает риск ИМ, как одного из исходов [43]. Этим данным можно доверять, поскольку они были получены в выборке численностью более 78 тыс. человек, которые участвовали в двух крупных исследованиях – ALLHAT и ASCOT [43]. В целом же информационная база по опыту клинического применения амлодипина малеата включает результаты примерно 800 клинических испытаний, в которых участвовало более 600 тыс. пациентов, подвергавшихся рандомизации [44].
Амлодипина безилат и амлодипина малеат
В большинстве европейских стран патент на амлодипин истек в 2004 г., в США он был продлен до 2007 г. Помимо оригинального бренда, защищенного патентом, во многих странах Европы (Германия, Швеция, Великобритания и т.д.) [45,46], в Корее [47] и Южной Африке [48] появились и генерические препараты, содержащие в качестве действующего вещества амлодипина малеат. Начиная с 2007 г., во всем мире стали доступны и генерики амлодипина безилата.
Изначально при производстве амлодипина использовалась соль малеиновой кислоты, однако позднее от нее отказались по ряду причин, в том числе из–за присущей лекарственному веществу нестабильности [7,34] и проблем с формовкой таблеток [34,39]. Также необходимо принимать во внимание и сведения о нефротоксическом действии малеиновой кислоты у грызунов [50,51]. В дальнейшем наличие токсического действия как у малеиновой кислоты, так и правадолина малеата подтвердилось в других исследованиях (см. выше) [30,52]. Таким образом, сомнения в безопасности коммерческих прописей амлодипина малеата у человека являются небезосновательными, что заставило провести ряд клинических испытаний перед тем, как начать его широкое применение на практике.
Сравнительное изучение биоэквивалентности амлодипина малеата проводилось в ходе лишь нескольких исследований, а опубликованы результаты только одного [17]. В свободном доступе имеются данные по эффективности и безопасности амлодипина малеата при эссенциальной гипертонии [47,53], но не стабильной стенокардии.
В процессе открытого рандомизированного двухэтапного перекрестного исследования в группе из 24 клинически здоровых добровольцев (возраст 24–45 лет) изучали, насколько биоэквивалентна однократная доза амлодипина малеата (Omicron Pharma) Норваску/амлодипина безилату (Pfizer) [17]. Поскольку не было выявлено никакой статистически достоверной разницы между этими веществами по показателям AUC и Cmax, а границы ДИ для амлодипина малеата (табл. 2) находились в пределах значений, допускаемых ЕАЛС (для Cmax 0,75–1,33), был сделан вывод об их биоэквивалентности. По–видимому, в клинической практике обе соли взаимозаменяемы, поскольку кинетика амлодипина малеата в плазме крови определяется только свойствами самой молекулы. Однако в США указанные лекарственные формы не будут считаться взаимозаменяемыми из–за того, что требования к препаратам с высокой вариабельностью здесь строже [14,15].
Учитывая тот факт, что равновесная концентрация амлодипина в крови должна быть гораздо выше [18], а гипертония чаще разивается в пожилом возрасте (показано, что у таких людей фармакокинетика амлодипина претерпевает изменения [54,55]), появляется веский аргумент в пользу необходимости оценивать биоэквивалентность в исследованиях с привлечением больных пожилого возраста, назначая лекарства в многократной дозе.
Эффективность и безопасность амлодипина безилата и амлодипина малеата анализировали в ходе двух многоцентровых рандомизированных контролируемых испытаний. Первое проводилось в течение 8 недель в Южной Корее (n=118), его цель заключалась в сравнительном изучении Норваска (Pfizer) и амлодипина малеата (производитель неизвестен) [47]. Второе исследование начиналось как двойное слепое (в течение 3 месяцев), а затем продолжилось как открытое (в течение 6 месяцев); польские ученые оценивали Норваск (Pfizer) и Тенокс (Krka, Словения) в группе из 250 человек [53]. В оба исследования были включены пациенты с артериальной гипертонией 2–3 ст. Вначале на протяжении двух недель принимавшиеся испытуемыми ранее лекарственные средства отменялись, а затем назначался амлодипина безилат или амлодипина малеат в дозе 5–10 мг 1 раз в сутки [47,53]. Согласно результатам корейского исследования по предварительно заданному критерию (изменение уровня диастолического АД на 4 мм рт.ст.) эффективность амлодипина малеата не превышала таковую амлодипина безилата (рис. 4) [47]. Однако выбор этой величины был произвольным и не подкрепленным четко установленными регулятивными нормами, а имеющиеся эпидемиологические данные заставляют считать, что такие колебания диастолического АД могут повлиять на кардиоваскулярные исходы [56]: если подвергнуть мета–анализу результаты 61 когортного исследования и 147 рандомизированных испытаний, то выяснится, что изменение диастолического АД на 4 мм рт.ст. обусловливает разницу по частоте коронарной болезни сердца на 20%, а инсульта – на 29% [57]. Более того, на фоне терапии амлодипина безилатом у несколько большего количества больных удавалось контролировать АД по сравнению с применением амлодипина малеата (92 и 86% соответственно). О сопоставимой эффективности обоих средств в качестве гипотензивных стало возможным говорить через 3 месяца, в течение которых велось исследование в Польше (рис. 4А) [53]. Хотя в последние 6 месяцев было отмечено статистически достоверное повышение АД на 0,9 мм рт.ст. в группе получавших амлодипина малеат (p<0,01 для диастолического АД и p<0,05 для систолического АД), значимого подъема уровня АД по сравнению с исходными величинами не отмечалось (рис. 4А) [53]. Оба препарата имеют сходный профиль безопасности (рис. 4Б) [47,53], но следует учесть, что это данные только за первые 3 месяца [47,53].
Отсутствие статистически достоверной разницы по результатам обоих исследований позволило их авторам сделать вывод, что амлодипина малеат может рассматриваться как альтернатива амлодипина безилату [47,53]. Однако не стоит делать торопливых обобщений и говорить о том, что эти лекарства взаимозаменяемы, ведь в этих исследованиях четко не определены критерии включения и исключения, численность выборок ограничена, нет отдаленных результатов (>3 месяцев). Чтобы говорить о терапевтической эквивалентности препаратов с точки зрения их гипотензивного действия, необходимы испытания в когортах численностью не менее 600 человек длительностью, как минимум, 6 месяцев. Ключевая задача лечения гипотензивными средствами – повлиять на фиксированные конечные точки (т.е. сократить частоту инсульта и ИМ), поэтому наилучшим решением при изучении терапевтической эквивалентности является проведение крупномасштабных продолжительных клинических исследований с непосредственным сравнением эффектов. Одно из таких длительных (примерно 4,4 года) исследований имело дизайн рандомизированного двойного слепого плацебо–контролируемого [58]. Но в опубликованном отчете содержатся данные только по совокупной частоте встречаемости основных кардиоваскулярных явлений во всех группах гипотензивной терапии по сравнению с плацебо, что исключает возможность независимой оценки амлодипина малеата. Также необходимо установить, является ли амлодипина малеат терапевтическим эквивалентом амлодипина безилата в качестве антиангинального средства.
Заключение
Несмотря на то, что терминология таких понятий, как биоэквивалентность и терапевтическая эквивалентность, была определена еще несколько десятилетий назад, до сих пор ведутся споры о взаимозаменяемости генериков и оригинальных препаратов. Биоэквивалентность, как указывают европейские и американские эксперты, подразумевает, но не гарантирует наличие терапевтической эквивалентности. Это может быть связано с целым рядом причин, в т.ч. с допускаемыми для генериков колебаниями в биоэквивалентности и преимущественной оценкой этих препаратов лишь в ходе кратковременных исследований с привлечением небольшого числа молодых и клинически здоровых лиц. Что еще более важно, отсутствие данных клинических испытаний с фиксированными конечными точками, которые свидетельствовали бы об эффективности и безопасности генериков в отдаленном периоде, ставит под сомнение значимость современных критериев в целом, поскольку пациент подвергается определенному риску.
Хотя генерики и оригинальные медикаменты должны содержать одни и те же активные компоненты, иметь одинаковый способ применения, обладать одной и той же силой, качеством, степенью очистки и фармакологической принадлежностью, они могут различаться, например, по составу примесей, которые должны быть инертными, но вовсе необязательно являются таковыми. Кроме того, несмотря на возможность упрощенной подачи заявки на регистрацию лекарства, в состав которого входит другая форма соли, изменение вида соли может повлиять на профиль препарата, о чем свидетельствует множество наблюдений (пример – оригинальный бренд амлодипина безилат и генерик амлодипина малеат). Хотя обе прописи, как показано, являются в соответствии с определением биоэквивалентными, до настоящего времени не проводилось их непосредственного сравнительного изучения в реальных клинических условиях на протяжении длительного срока. Более того, необоснованного назначения препаратов амлодипина малеата следует избегать из–за (потенциальной) нефротоксичности малеиновой кислоты/малеатов у животных и/или присутствия в лекарстве биологически активных примесей, появляющихся вследствие разрушения действующего вещества или иных процессов. Необходимо провести еще ряд исследований с тем, чтобы с полной уверенностью говорить о терапевтической взаимозаменяемости безилатной и малеиновой соли амлодипина.
* Норваск, Истин и Амлор являются зарегистрированными тор-
говыми марками компании Pfizer




Реферат подготовлен к.м.н. Е.Б. Третьяк
по материалам статьи P.A. Meredith
«Potential concerns about generic substitution: bioequivalence versus therapeutic equivalence
of different amlodipine salt forms»
Current Medical Research and Opinion 2009;
Vol. 25, № 9: 2179–2189
по материалам статьи P.A. Meredith
«Potential concerns about generic substitution: bioequivalence versus therapeutic equivalence
of different amlodipine salt forms»
Current Medical Research and Opinion 2009;
Vol. 25, № 9: 2179–2189
Литература
1. Genazzani AA, Pattarino F. Difficulties in the production of identical drug products from a pharmaceutical technology viewpoint. Drugs R D 2008;9:65-72.
2. World Health Organization. Available at: http://www.who.int/ trade/glossary/story034/en/index.html.
3. U.S. Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Orange Book – Approved drug products with therapeutic equivalence evaluations, 28th edn, 2008. Available at: http://www.fda.gov/cder/orange/obannual.pdf.
4. European Medicines Agency (EMEA), Committee for Proprietary Medicinal Products (CPMP), Note for guidance on the investigation of bioavailability and bioequivalence, CPMP/EWP/QWP/1401/98, July 2001.
5. U.S. Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Guidance for industry: Bioavailability and bioequivalence studies for orally administered drug products – general considerations, March 2003.
6. Meredith PA, The Unique Adalat Story – Nifedipine Gastrointestinal Therapeutic System. European Cardiovascular Disease 2007; Issue 1, July 2007. Available at: http://www.touchbriefings.com/cdps/cditem.cfm?nid?2744&cid?5#Hypertension.
7. Verbeeck RK, Kanfer I, Walker RB. Generic substitution: the use of medicinal products containing different salts and implications for safety and efficacy. Eur J Pharm Sci 2006;28:1-6.
8. Basak AK, Raw AS, Al Hakim AH, et al. Pharmaceutical impurities: regulatory perspective for Abbreviated New Drug Applications. Adv Drug Deliv Rev 2007;59:64-72.
9. Chen ML, Shah V, Patnaik R, et al. Bioavailability and bioequivalence: an FDA regulatory overview. Pharm Res 2001;18:1645-50.
10. Nakai K, Fujita M, Ogata H. International harmonization of bioequivalence studies and issues shared in common. Yakugaku Zasshi 2000;120:1193-200.
11. Meredith P. Bioequivalence and other unresolved issues in generic drug substitution. Clin Ther 2003;25:2875-90.
12. Birkett DJ. Generics – equal or not? Austr Prescr 2003;26:85-7.
13. Besag FM. Is generic prescribing acceptable in epilepsy? Drug Saf 2000;23:173-82.
14. European Medicines Agency (EMEA), CPMP Efficacy Working Party Therapeutic Subgroup on Pharmacokinetics (EWP-PK), July 2006. Available at: http://www.emea.europa.eu/pdfs/human/ewp/4032606en.pdf.
15. Midha KK, Rawson MJ, Hubbard JW. The bioequivalence of highly variable drugs and drug products. Int J Clin Pharmacol Ther 2005;43:485-98.
16. U.S. Food and Drug Administration (FDA), Therapeutic equivalence of generic drugs: Letter to health practitioners, January 1998. Available at: http://www.fda.gov/cder/news/nightgenlett.htm.
17. Mignini F, Tomassoni D, Traini E, et al. Single-dose, randomized, crossover bioequivalence study of amlodipine maleate versus amlodipine besylate in healthy volunteers. Clin Exp Hypertens 2007;29:539-52.
18. Donnelly R, Meredith PA, Miller SH, et al. Pharmacodynamic modeling of the antihypertensive response to amlodipine. Clin Pharmacol Ther 1993;54:303-10.
19. Meredith PA. Generic drugs. Therapeutic equivalence. Drug Saf 1996;15:233-42.
20. Carter BL, Noyes MA, Demmler RW. Differences in serum concentrations of and responses to generic verapamil in the elderly. Pharmacotherapy 1993;13:359-68.
21. Borgherini G. The bioequivalence and therapeutic efficacy of generic versus brand-name psychoactive drugs. Clin Ther 2003;25:1578-92.
22. Nakai K, Fujita M, Ogata H. New bioequivalence studies: individual bioequivalence and population bioequivalence. Yakugaku Zasshi 2000;120:1201-8.
23. Chen ML, Lesko LJ. Individual bioequivalence revisited. Clin Pharmacokinet 2001;40:701-6.
24. Laroche ML, Merle L. Generic and brand-name drugs. Are different criteria sufficiently taken into account before granting market authorisation? Acta Clin Belg Suppl 2006;(1):48-50.
25. Furberg BD, Furberg CD. Are all drugs of a class interchangeable? In: Evaluating Clinical Research: All that Glitters is not Gold. New York: Springer, 2007:115-19.
26. Wiecek A, Mikhail A. European regulatory guidelines for biosimilars. Nephrol Dial Transplant 2006;21(Suppl 5):v17-20.
27. Davies G. Changing the salt, changing the drug. Pharm J 2001;266:322-3.
28. Frank RG. The ongoing regulation of generic drugs. N Engl J Med 2007;357:1993-6.
29. Snider DA, Addicks W, Owens W. Polymorphism in generic drug product development. Adv Drug Deliv Rev 2004;56:391-5.
30. Everett RM, Descotes G, Rollin M, et al. Nephrotoxicity of pravadoline maleate (WIN 48098-6) in dogs: evidence of maleic acid-induced acute tubular necrosis. Fundam Appl Toxicol 1993;21:59-65.
31. Olovson SG, Havu N, Rega°rdh CG, et al. Oesophageal ulcerations and plasma levels of different alprenolol salts: potential implications for the clinic. Acta Pharmacol Toxicol (Copenh) 1986;58:55-60.
32. U.S. Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Guidance for industry:Q3A impurities in new drug substances, June 2008.
33. U.S. Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Guidance for industry:Q3B(R2) impurities in new drug products, July 2006.
34. Amlodipine Citizen Petition by Pfizer Inc. Available at: http://www.fda.gov/ohrms/dockets/dailys/03/Sept03/090303/03p-0408-cp00001-08-Tab-G-vol3.pdf.
35. Sudhakar P, Nirmala M, Moses Babu J, et al. Identification and characterization of potential impurities of amlodipine maleate. J Pharm Biomed Anal 2006;40:605-13.
36. Murakami T, Fukutsu N, Kondo J, et al. Application of liquid chromatography-two-dimensional nuclear magnetic resonance spectroscopy using pre-concentration column trapping and liquid chromatography-mass spectrometry for the identification of degradation products in stressed commercial amlodipine maleate tablets. J Chromatogr A 2008;1181:67-76.
37. European Medicines Agency (EMEA), Committee for Medicinal Products for Human Use (CHMP), Guideline on the limits of genotoxic impurities, EMEA/CHMP/QWP/251344/2006, London 2006.
38. Murdoch D, Heel RC. Amlodipine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in cardiovascular disease. Drugs 1991;41:478-505.
39. Norvasc. Pfizer Inc. Available at: http://www.fda.gov/cder/foi/label/2007/019787s042lbl.pdf
40. Clavijo GA, de Clavijo IV, Weart CW. Amlodipine: a new calcium antagonist. Am J Hosp Pharm 1994;51:59-68.
41. Istin. Pfizer Inc. Available at: http://emc.medicines.org.uk/emc/assets/c/ html/displaydoc.asp?documentid?1466.
42. Amlor. Pfizer Inc. Available at: http://www.pfizer.fr/Portals/0/AMLOR%20Notice%20commune%205%20et%2010mg%20INT.pdf.
43. Wang JG, Li Y, Franklin SS, et al. Prevention of stroke and myocardial infarction by amlodipine and angiotensin receptor blockers: a quantitative overview. Hypertension 2007;50:181-8.
44. Data on file. Pfizer Inc.
45. European Medicines Agency (EMEA), Committee for Proprietary Medicinal Products (CPMP), Opinion following an article 29 referral: Amlovita. London 26 April 2004. EMEA/CPMP/539/04.
46. European Medicines Agency (EMEA), Committee for Proprietary Medicinal Products (CPMP), Opinion following an article 29 referral: Talam. London 27 April 2004. EMEA/CPMP/540/04.
47. Park S, Chung N, Kwon J, et al. Results of a multicenter, 8-week, parallel-group, randomized, double-blind, doubledummy, phase III clinical trial to evaluate the efficacy and tolerability of amlodipine maleate versus amlodipine besylate in Korean patients with mild to moderate hypertension. Clin Ther 2005;27:441-50.
48. New amlodipine (Amloc) product available in South Africa. Cardiovasc J S Afr 2005;16:61.
49. Pragai G, Orosz E, Szilagyi J, et al. United States Patent Application Publication, US 2005/0019395 A1, January 27,2005.
50. Harrison HE, Harrison HC. Experimental production of renal glycosuria, phosphaturia, and aminoaciduria by injection of maleic acid. Science 1954;120:606-8.
51. Angielsky S, Rogulski J. Aminoaciduria caused by maleic acid. III. The effect of sulfhydryl compounds. Acta Biochim Pol 1959;6:411-15.
52. Zager RA, Johnson AC, Naito M, et al. Maleate nephrotoxicity: mechanisms of injury and correlates with ischemic/hypoxic tubular cell death. Am J Physiol Renal Physiol 2008; 294:F187-97.
53. Makowiecka-Cies?la M, Januszewicz A, Prejbisz A, et al. Nine month follow-up of amlodipine maleate and amlodipine besylate treatment in patients with essential hypertension: does the salt form matter? Arterial Hypertension 2005;9:364-73.
54. Elliott HL, Meredith PA, Reid JL, et al A comparison of the disposition of single oral doses of amlodipine in young and elderly subjects. J Cardiovasc Pharmacol 1988;12 (Suppl 7):S64-6.
55. Abernethy DR, Gutkowska J, Winterbottom LM. Effects of amlodipine, a long-acting dihydropyridine calcium antagonist in aging hypertension: pharmacodynamics in relation to disposition. Clin Pharmacol Ther 1990;48:76-86.
56. Verdecchia P, Reboldi G, Angeli F, et al. Angiotensin-converting enzyme inhibitors and calcium channel blockers for coronary heart disease and stroke prevention. Hypertension. 2005;46:386-92.
57. Wald DS, Law M, Morris JK, et al. Combination therapy versus monotherapy in reducing blood pressure: meta-analysis on 11,000 participants from 42 trials. Am J Med 2009;122:290-300.
58. Neaton JD, Grimm Jr RH, Prineas RJ, et al. Treatment of Mild Hypertension Study. Final results. JAMA 1993;270:713-24.
59. U.S. Food and Drug Administration (FDA), Department of Health and Human Services, Food and Drug Administration Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER): Guidance for Industry: Population Pharmacokinetics, February 1999. Available at: http://www.fda.gov/CDER/guidance/1852fnl.pdf



