




–¶–µ–ї—М—О –і–∞–љ–љ–Њ–є —А–∞–±–Њ—В—Л —П–≤–Є–ї–Њ—Б—М –Є–Ј—Г—З–µ–љ–Є–µ —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є–Ї–Є –њ–Њ—А–Њ—И–Ї–∞ –Є —А–∞—Б—В–≤–Њ—А–∞ —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є —Д–Њ—А–Љ—Л –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –њ–Њ—Б–ї–µ –Њ–і–љ–Њ–Ї—А–∞—В–љ–Њ–≥–Њ –њ–µ—А–Њ—А–∞–ї—М–љ–Њ–≥–Њ –≤–≤–µ–і–µ–љ–Є—П –Ї—А—Л—Б–∞–Љ.
–Ь–µ—В–Њ–і—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П
–≠–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –ґ–Є–≤–Њ—В–љ—Л–µ
–Є —Е–Є—А—Г—А–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—Ж–µ–і—Г—А—Л
–Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –≤—Л–њ–Њ–ї–љ–µ–љ–Њ –љ–∞ —Б–∞–Љ—Ж–∞—Е –Ї—А—Л—Б –ї–Є–љ–Є–Є Wistar –Љ–∞—Б—Б–Њ–є 250–300 –≥. –Ц–Є–≤–Њ—В–љ—Л–Љ –≤ —Б—В–µ—А–Є–ї—М–љ—Л—Е —Г—Б–ї–Њ–≤–Є—П—Е –њ–Њ–і –љ–∞—А–Ї–Њ–Ј–Њ–Љ (–њ–µ–љ—В–Њ–±–∞—А–±–Є—В–∞–ї, 50 –Љ–≥/–Ї–≥ –≤–љ—Г—В—А–Є–±—А—О—И–Є–љ–љ–Њ) –Є–Љ–њ–ї–∞–љ—В–Є—А–Њ–≤–∞–ї–Є –Ї–∞—В–µ—В–µ—А –≤ –±–µ–і—А–µ–љ–љ—Г—О –≤–µ–љ—Г –і–ї—П –њ–Њ—Б–ї–µ–і—Г—О—Й–µ–≥–Њ –Њ—В–±–Њ—А–∞ –Њ–±—А–∞–Ј—Ж–Њ–≤ –Ї—А–Њ–≤–Є. –І–µ—А–µ–Ј —Б—Г—В–Ї–Є –њ–Њ—Б–ї–µ –Њ–њ–µ—А–∞—Ж–Є–Є –±–Њ–і—А—Б—В–≤—Г—О—Й–Є–Љ –ґ–Є–≤–Њ—В–љ—Л–Љ –љ–∞—В–Њ—Й–∞–Ї —З–µ—А–µ–Ј –Ј–Њ–љ–і –≤–≤–Њ–і–Є–ї–Є –≤ –ґ–µ–ї—Г–і–Њ–Ї –Ї–Њ—Н–љ–Ј–Є–Љ Q10 –≤ –і–Њ–Ј–µ 10 –Љ–≥/–Ї–≥: –Ї—А—Л—Б–∞–Љ –њ–µ—А–≤–Њ–є –≥—А—Г–њ–њ—Л (n=7) – —А–∞—Б—В–≤–Њ—А —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 («–Ъ—Г–і–µ—Б–∞–љ», –Ч–Р–Ю «–Р–Ъ–Т–Ш–Ю–Э», –Ь–Њ—Б–Ї–≤–∞, –†–Њ—Б—Б–Є—П), –Ї—А—Л—Б–∞–Љ –≤—В–Њ—А–Њ–є –≥—А—Г–њ–њ—Л (n=7) – –њ–Њ—А–Њ—И–Њ–Ї –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –≤ –≤–Є–і–µ –≤–Ј–≤–µ—Б–Є –≤ 0,2% —А–∞—Б—В–≤–Њ—А–µ –Љ–µ—В–Є–ї—Ж–µ–ї–ї—О–ї–Њ–Ј—Л.
–Ю—В–±–Њ—А –њ—А–Њ–± –Ї—А–Њ–≤–Є (–њ–Њ 0,3 –Љ–ї) –њ—А–Њ–≤–Њ–і–Є–ї–Є –і–Њ –≤–≤–µ–і–µ–љ–Є—П –њ—А–µ–њ–∞—А–∞—В–∞ –Є —Б–њ—Г—Б—В—П 2, 3, 4, 5, 7, 9, 24, 36 –Є 48 —З –њ–Њ—Б–ї–µ –≤–≤–µ–і–µ–љ–Є—П. –Ю–±—А–∞–Ј—Ж—Л –Ї—А–Њ–≤–Є —Ж–µ–љ—В—А–Є—Д—Г–≥–Є—А–Њ–≤–∞–ї–Є, –њ–ї–∞–Ј–Љ—Г –Ї—А–Њ–≤–Є –Њ—В–±–Є—А–∞–ї–Є, –Ј–∞–Љ–Њ—А–∞–ґ–Є–≤–∞–ї–Є –Є —Е—А–∞–љ–Є–ї–Є –њ—А–Є –20°–° –і–Њ –њ—А–Њ–≤–µ–і–µ–љ–Є—П –Ї–Њ–ї–Є—З–µ—Б—В–≤–µ–љ–љ–Њ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞ –Ї–Њ—Н–љ–Ј–Є–Љ–Њ–≤ Q. –Э–∞ –њ—А–Њ—В—П–ґ–µ–љ–Є–Є –і–≤—Г—Е —Б—Г—В–Њ–Ї –њ–Њ—Б–ї–µ –≤–≤–µ–і–µ–љ–Є—П –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –ґ–Є–≤–Њ—В–љ—Л—Е –љ–µ –Ї–Њ—А–Љ–Є–ї–Є, –љ–µ –Њ–≥—А–∞–љ–Є—З–Є–≤–∞—П –і–Њ—Б—В—Г–њ –Ї –≤–Њ–і–µ.
–Ю–њ—А–µ–і–µ–ї–µ–љ–Є–µ –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10
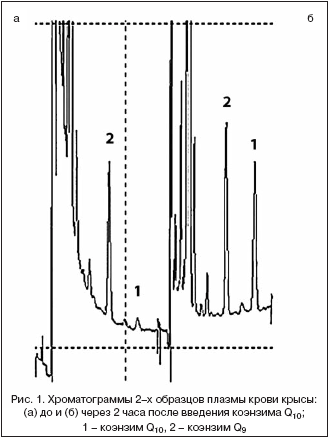
–Ъ–Њ–ї–Є—З–µ—Б—В–≤–µ–љ–љ—Л–є –∞–љ–∞–ї–Є–Ј –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –≤ –њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є –њ—А–Њ–≤–Њ–і–Є–ї–Є –њ–Њ –Љ–µ—В–Њ–і–Є–Ї–µ [5] —Б –љ–µ–Ї–Њ—В–Њ—А—Л–Љ–Є –Љ–Њ–і–Є—Д–Є–Ї–∞—Ж–Є—П–Љ–Є. –Ъ 100 –Љ–Ї–ї –њ–ї–∞–Ј–Љ—Л –Ї—А–Њ–≤–Є –і–Њ–±–∞–≤–ї—П–ї–Є 220 –Љ–Ї–ї —Н—В–∞–љ–Њ–ї–∞ –Є 550 –Љ–Ї–ї n––≥–µ–Ї—Б–∞–љ–∞, —В—Й–∞—В–µ–ї—М–љ–Њ –≤—Б—В—А—П—Е–Є–≤–∞–ї–Є –≤ —В–µ—З–µ–љ–Є–µ 10 –Љ–Є–љ, —Ж–µ–љ—В—А–Є—Д—Г–≥–Є—А–Њ–≤–∞–ї–Є –њ—А–Є 3000 –Њ–±/–Љ–Є–љ –≤ —В–µ—З–µ–љ–Є–µ 3 –Љ–Є–љ –Є –≤–µ—А—Е–љ–Є–є —Б–ї–Њ–є n––≥–µ–Ї—Б–∞–љ–∞ –Њ—В–±–Є—А–∞–ї–Є –≤ –Њ–±—К–µ–Љ–µ 500 –Љ–Ї–ї. –Ъ –Њ—Б—В–∞—В–Ї—Г –і–Њ–±–∞–≤–ї—П–ї–Є –µ—Й–µ 550 –Љ–Ї–ї n––≥–µ–Ї—Б–∞–љ–∞ –Є –њ–Њ–≤—В–Њ—А—П–ї–Є –њ—А–Њ—Ж–µ–і—Г—А—Г —Н–Ї—Б—В—А–∞–Ї—Ж–Є–Є –Є –Њ—В–±–Њ—А–∞ —Н–Ї—Б—В—А–∞–Ї—В–∞. –Ю–±—К–µ–і–Є–љ–µ–љ–љ—Л–є —Н–Ї—Б—В—А–∞–Ї—В —Г–њ–∞—А–Є–≤–∞–ї–Є –і–Њ—Б—Г—Е–∞, —А–∞—Б—В–≤–Њ—А—П–ї–Є –≤ 100 –Љ–Ї–ї —Н—В–∞–љ–Њ–ї–∞ –Є –≤–Њ—Б—Б—В–∞–љ–∞–≤–ї–Є–≤–∞–ї–Є –Њ–Ї–Є—Б–ї–µ–љ–љ—Г—О —Д–Њ—А–Љ—Г –Ї–Њ—Н–љ–Ј–Є–Љ–Њ–≤ Q9 –Є Q10 –і–Њ–±–∞–≤–ї–µ–љ–Є–µ–Љ 10 –Љ–Ї–ї 5% —А–∞—Б—В–≤–Њ—А–∞ –љ–∞—В—А–Є—П —В–µ—В—А–∞–≥–Є–і—А–Њ–±–Њ—А–∞—В–∞ –≤ —Н—В–∞–љ–Њ–ї–µ. 10 –Љ–Ї–ї –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–љ–Њ–≥–Њ —Н–Ї—Б—В—А–∞–Ї—В–∞ –∞–љ–∞–ї–Є–Ј–Є—А–Њ–≤–∞–ї–Є —Б –њ–Њ–Љ–Њ—Й—М—О –≤—Л—Б–Њ–Ї–Њ—Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ–є –ґ–Є–і–Ї–Њ—Б—В–љ–Њ–є —Е—А–Њ–Љ–∞—В–Њ–≥—А–∞—Д–Є–Є —Б —Н–ї–µ–Ї—В—А–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є–Љ –і–µ—В–µ–Ї—В–Є—А–Њ–≤–∞–љ–Є–µ–Љ –љ–∞ –Њ–±–Њ—А—Г–і–Њ–≤–∞–љ–Є–Є —Д–Є—А–Љ—Л «Environmental Sciences Associate, Inc.» (–°–®–Р) – –љ–∞—Б–Њ—Б –Љ–Њ–і–µ–ї–Є 580 –Є —Н–ї–µ–Ї—В—А–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є–є –і–µ—В–µ–Ї—В–Њ—А «Coulochem II». –£—Б–ї–Њ–≤–Є—П –њ—А–Њ–≤–µ–і–µ–љ–Є—П —Е—А–Њ–Љ–∞—В–Њ–≥—А–∞—Д–Є—З–µ—Б–Ї–Њ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞ –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–ї–Є —А–∞–Ј–і–µ–ї–µ–љ–Є–µ –Ї–Њ—Н–љ–Ј–Є–Љ–Њ–≤ Q9 –Є Q10, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ–µ –≤–≤–Є–і—Г —В–Њ–≥–Њ, —З—В–Њ –і–Њ–Љ–Є–љ–Є—А—Г—О—Й–µ–є —Д–Њ—А–Љ–Њ–є –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q —Г –Ї—А—Л—Б —П–≤–ї—П–µ—В—Б—П –Ї–Њ—Н–љ–Ј–Є–Љ Q9 (—А–Є—Б. 1). –†–∞–Ј–і–µ–ї–µ–љ–Є–µ –њ—А–Њ–≤–Њ–і–Є–ї–Є –≤ –Є–Ј–Њ–Ї—А–∞—В–Є—З–µ—Б–Ї–Њ–Љ —А–µ–ґ–Є–Љ–µ –љ–∞ –Ї–Њ–ї–Њ–љ–Ї–µ 150—Е4,6 –Љ–Љ —Б —Б–Њ—А–±–µ–љ—В–Њ–Љ –°18 (5 –Љ–Ї–Љ) –њ—А–Є —Б–Ї–Њ—А–Њ—Б—В–Є –њ–Њ—В–Њ–Ї–∞ —Н–ї—О—Н–љ—В–∞ 1,3 –Љ–ї/–Љ–Є–љ. –Я–Њ–і–≤–Є–ґ–љ–∞—П —Д–∞–Ј–∞ —Б–Њ–і–µ—А–ґ–∞–ї–∞ 0,3% NaCl –≤ —Б–Љ–µ—Б–Є —Н—В–∞–љ–Њ–ї: –Љ–µ—В–∞–љ–Њ–ї: 7% HClO4 (970:20:10). –≠–ї–µ–Ї—В—А–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Њ–µ –і–µ—В–µ–Ї—В–Є—А–Њ–≤–∞–љ–Є–µ –Њ—Б—Г—Й–µ—Б—В–≤–ї—П–ї–Є —Б –њ–Њ–Љ–Њ—Й—М—О –∞–љ–∞–ї–Є—В–Є—З–µ—Б–Ї–Њ–є —П—З–µ–є–Ї–Є (model 5011) –њ—А–Є –љ–∞–њ—А—П–ґ–µ–љ–Є–Є –љ–∞ –њ–µ—А–≤–Њ–є –њ–∞—А–µ —Н–ї–µ–Ї—В—А–Њ–і–Њ–≤ –50 –Љ–Т –Є +350 –Љ–Т – –љ–∞ –≤—В–Њ—А–Њ–є. –Т—А–µ–Љ—П —Г–і–µ—А–ґ–Є–≤–∞–љ–Є—П —Б–Њ—Б—В–∞–≤–Є–ї–Њ –і–ї—П –Ї–Њ—Н–љ–Ј–Є–Љ–Њ–≤ Q9 –Є Q10 – 6,5 –Є 8,5 –Љ–Є–љ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ. –†–µ–≥–Є—Б—В—А–∞—Ж–Є—О –Є –Њ–±—А–∞–±–Њ—В–Ї—Г —Е—А–Њ–Љ–∞—В–Њ–≥—А–∞—Д–Є—З–µ—Б–Ї–Є—Е –і–∞–љ–љ—Л—Е –њ—А–Њ–≤–Њ–і–Є–ї–Є —Б –њ–Њ–Љ–Њ—Й—М—О –Ї–Њ–Љ–њ—М—О—В–µ—А–љ–Њ–є –њ—А–Њ–≥—А–∞–Љ–Љ—Л —Д–Є—А–Љ—Л «Environmental Sciences Associate, Inc.» (–°–®–Р).
–Ю–њ—А–µ–і–µ–ї–µ–љ–Є–µ —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Є—Е —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10
–Ф–ї—П –њ–Њ—А–Њ—И–Ї–∞ –Є —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є —Д–Њ—А–Љ—Л –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –±—Л–ї–Є –Њ–њ—А–µ–і–µ–ї–µ–љ—Л –Є —А–∞—Б—Б—З–Є—В–∞–љ—Л —Б–ї–µ–і—Г—О—Й–Є–µ –њ–∞—А–∞–Љ–µ—В—А—Л —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є–Ї–Є: Tmax – –≤—А–µ–Љ—П –і–Њ—Б—В–Є–ґ–µ–љ–Є—П –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є, Cmax – –≤–µ–ї–Є—З–Є–љ–∞ –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є, AUCo–t – –њ–ї–Њ—Й–∞–і—М –њ–Њ–і —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Њ–є –Ї—А–Є–≤–Њ–є “–Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П––≤—А–µ–Љ—П”, Cmax/AUCo–t – –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–∞—П —Б–Ї–Њ—А–Њ—Б—В—М –≤—Б–∞—Б—Л–≤–∞–љ–Є—П. –Ф–ї—П —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є —Д–Њ—А–Љ—Л —А–∞—Б—Б—З–Є—В—Л–≤–∞–ї–Є: f – –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ—Г—О –±–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В—М –Є—Б—Б–ї–µ–і—Г–µ–Љ–Њ–є –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ–Њ–є —Д–Њ—А–Љ—Л –њ–Њ –Њ—В–љ–Њ—И–µ–љ–Є—О –Ї —Б—А–∞–≤–љ–Є–≤–∞–µ–Љ–Њ–є, –Њ–њ—А–µ–і–µ–ї—П–µ–Љ—Г—О –Њ—В–љ–Њ—И–µ–љ–Є–µ–Љ AUCo–t,T/AUCo–t,R, f’ – –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ—Г—О —Б—В–µ–њ–µ–љ—М –≤—Б–∞—Б—Л–≤–∞–љ–Є—П, –Њ–њ—А–µ–і–µ–ї—П–µ–Љ—Г—О –Њ—В–љ–Њ—И–µ–љ–Є–µ–Љ Cmax,T/Cmax,R.
–Ч–љ–∞—З–µ–љ–Є—П –њ–∞—А–∞–Љ–µ—В—А–Њ–≤ —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є–Ї–Є –±—Л–ї–Є —А–∞—Б—Б—З–Є—В–∞–љ—Л –Љ–Њ–і–µ–ї—М–љ–Њ––љ–µ–Ј–∞–≤–Є—Б–Є–Љ—Л–Љ–Є –Љ–µ—В–Њ–і–∞–Љ–Є. –Т–µ–ї–Є—З–Є–љ—Г Cmax –Є –≤—А–µ–Љ—П –µ–µ –і–Њ—Б—В–Є–ґ–µ–љ–Є—П –Њ–њ—А–µ–і–µ–ї—П–ї–Є –Є–Ј —Д–∞–Ї—В–Є—З–µ—Б–Ї–Є—Е –Ј–љ–∞—З–µ–љ–Є–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–є. –Я–ї–Њ—Й–∞–і—М –њ–Њ–і —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Њ–є –Ї—А–Є–≤–Њ–є (AUCo–t) —А–∞—Б—Б—З–Є—В—Л–≤–∞–ї–Є –Љ–µ—В–Њ–і–Њ–Љ —В—А–∞–њ–µ—Ж–Є–є.
–†–∞—Б—З–µ—В –±–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В–Є –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –њ—А–Њ–Є–Ј–≤–Њ–і–Є–ї—Б—П –Љ–µ—В–Њ–і–Њ–Љ —Б—А–∞–≤–љ–µ–љ–Є—П –њ–ї–Њ—Й–∞–і–µ–є –њ–Њ–і –Ї—А–Є–≤—Л–Љ–Є –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –≤ –њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є –Њ—В –≤—А–µ–Љ–µ–љ–Є, –њ–Њ—Б—В—А–Њ–µ–љ–љ—Л–Љ–Є –і–ї—П –і–≤—Г—Е –≥—А—Г–њ–њ –Ї—А—Л—Б.
–Ю–±—А–∞–±–Њ—В–Ї–∞ —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤. –†–µ–Ј—Г–ї—М—В–∞—В—Л –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ—Л –Ї–∞–Ї —Б—А–µ–і–љ–µ–µ –Ј–љ–∞—З–µ–љ–Є–µ ± —Б—В–∞–љ–і–∞—А—В–љ–Њ–µ –Њ—В–Ї–ї–Њ–љ–µ–љ–Є–µ (M±m). –Ь–µ–ґ–≥—А—Г–њ–њ–Њ–≤—Л–µ —А–∞–Ј–ї–Є—З–Є—П –Њ—Ж–µ–љ–Є–≤–∞–ї–Є, –Є—Б–њ–Њ–ї—М–Ј—Г—П t–test. –†–∞–Ј–ї–Є—З–Є—П —Б—З–Є—В–∞–ї–Є –Ј–љ–∞—З–Є–Љ—Л–Љ–Є –њ—А–Є –≤–µ—А–Њ—П—В–љ–Њ—Б—В–Є –Њ—И–Є–±–Ї–Є –Љ–µ–љ–µ–µ 5% (p<0,05).
–†–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П
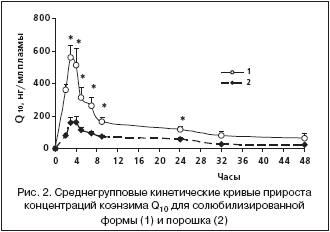
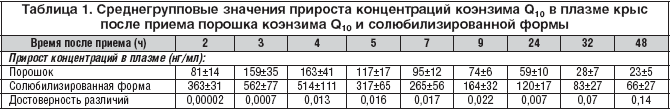
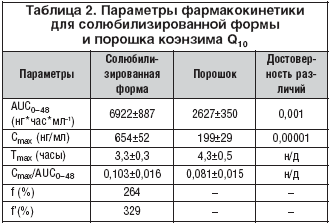
–Я–Њ–ї—Г—З–µ–љ–љ—Л–µ –≤–µ–ї–Є—З–Є–љ—Л –≤—А–µ–Љ–µ–љ–Є –і–Њ—Б—В–Є–ґ–µ–љ–Є—П –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –≤ –њ–ї–∞–Ј–Љ–µ (3–4 —З) —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ –Љ–µ–і–ї–µ–љ–љ–Њ–є –∞–±—Б–Њ—А–±—Ж–Є–Є –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –≤ –ґ–µ–ї—Г–і–Њ—З–љ–Њ––Ї–Є—И–µ—З–љ–Њ–Љ —В—А–∞–Ї—В–µ –≤—Б–ї–µ–і—Б—В–≤–Є–µ –µ–≥–Њ –≥–Є–і—А–Њ—Д–Њ–±–љ–Њ—Б—В–Є –Є –±–Њ–ї—М—И–Њ–≥–Њ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ–≥–Њ –≤–µ—Б–∞. –Т—А–µ–Љ—П –і–Њ—Б—В–Є–ґ–µ–љ–Є—П –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Є –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–∞—П —Б–Ї–Њ—А–Њ—Б—В—М –≤—Б–∞—Б—Л–≤–∞–љ–Є—П –њ–Њ—А–Њ—И–Ї–∞ –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –Є —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є —Д–Њ—А–Љ—Л –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ –љ–µ —А–∞–Ј–ї–Є—З–∞–ї–Є—Б—М. –Ф—А—Г–≥–Є–µ —Д–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –њ–∞—А–∞–Љ–µ—В—А—Л –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –≤ —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є —Д–Њ—А–Љ–µ —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –Њ—В–ї–Є—З–∞–ї–Є—Б—М –Њ—В —В–∞–Ї–Њ–≤—Л—Е –≤ –њ–Њ—А–Њ—И–Ї–µ (—А–Є—Б. 2). –°—А–µ–і–љ–Є–µ –Ј–љ–∞—З–µ–љ–Є—П –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–є –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –≤ –њ–ї–∞–Ј–Љ–µ –≤ —В–µ—З–µ–љ–Є–µ —Б—Г—В–Њ–Ї –њ–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞ —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є —Д–Њ—А–Љ—Л –±—Л–ї–Є –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ –≤—Л—И–µ, —З–µ–Љ –њ–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞ –њ–Њ—А–Њ—И–Ї–∞ (—В–∞–±–ї. 1). –Ь–∞–Ї—Б–Є–Љ–∞–ї—М–љ–∞—П –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –≤ –њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є –њ–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞ —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є —Д–Њ—А–Љ—Л –±—Л–ї–∞ –њ—А–Є–Љ–µ—А–љ–Њ –≤ 3,3 —А–∞–Ј–∞ –≤—Л—И–µ, –∞ –њ–ї–Њ—Й–∞–і—М –њ–Њ–і –Ї—А–Є–≤–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П––≤—А–µ–Љ—П – –≤ 2,6 —А–∞–Ј–∞ –±–Њ–ї—М—И–µ –њ–Њ —Б—А–∞–≤–љ–µ–љ–Є—О —Б –њ–Њ—А–Њ—И–Ї–Њ–Љ (—В–∞–±–ї. 2). –°–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ, –±–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В—М –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –≤ —Б–Њ—Б—В–∞–≤–µ —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є —Д–Њ—А–Љ—Л –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ –њ–Њ—А–Њ—И–Ї–∞ —Б–Њ—Б—В–∞–≤–ї—П–ї–∞ 264%, –∞ –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–∞—П —Б—В–µ–њ–µ–љ—М –≤—Б–∞—Б—Л–≤–∞–љ–Є—П 329% (—В–∞–±–ї. 2).
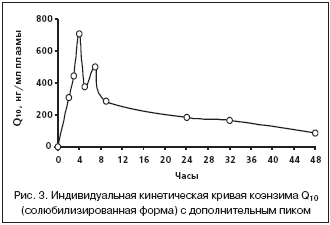
–Ъ–Є–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –Ї—А–Є–≤—Л–µ –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 —Г –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е –ґ–Є–≤–Њ—В–љ—Л—Е –Є–Љ–µ–ї–Є –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л–µ –њ–Є–Ї–Є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–є, –љ–∞–±–ї—О–і–∞–≤—И–Є–µ—Б—П –≤ –Є–љ—В–µ—А–≤–∞–ї–µ –Њ—В 7 –і–Њ 24 —З–∞—Б–Њ–≤ –њ–Њ—Б–ї–µ –≤–≤–µ–і–µ–љ–Є—П –≤–µ—Й–µ—Б—В–≤–∞ (—А–Є—Б. 3). –Ш–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –ґ–Є—А–Њ—А–∞—Б—В–≤–Њ—А–Є–Љ—Л–є –Ї–Њ—Н–љ–Ј–Є–Љ Q10 –њ–Њ—Б–ї–µ –≤—Б–∞—Б—Л–≤–∞–љ–Є—П –≤ –Ї–Є—И–µ—З–љ–Є–Ї–µ –Ј–∞—Е–≤–∞—В—Л–≤–∞–µ—В—Б—П –Є–Ј —Ж–Є—А–Ї—Г–ї—П—Ж–Є–Є —В–Ї–∞–љ—П–Љ–Є –њ–µ—З–µ–љ–Є [3,13]. –Я–Њ––≤–Є–і–Є–Љ–Њ–Љ—Г, –љ–∞–ї–Є—З–Є–µ –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л—Е –њ–Є–Ї–Њ–≤ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–є –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10, –љ–∞–±–ї—О–і–∞–≤—И–µ–µ—Б—П —В–∞–Ї–ґ–µ –≤ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –љ–∞ –ї—О–і—П—Е [10] –Є –љ–∞ –Љ–Њ—А—Б–Ї–Є—Е —Б–≤–Є–љ–Ї–∞—Е [11], —П–≤–ї—П–µ—В—Б—П –Њ—В—А–∞–ґ–µ–љ–Є–µ–Љ —Д–µ–љ–Њ–Љ–µ–љ–∞ —Н–љ—В–µ—А–Њ–≥–µ–њ–∞—В–Є—З–µ—Б–Ї–Њ–є —Ж–Є—А–Ї—Г–ї—П—Ж–Є–Є. –≠–њ–Є–Ј–Њ–і—Л –њ–Њ–≤—В–Њ—А–љ–Њ–≥–Њ –≤—Б–∞—Б—Л–≤–∞–љ–Є—П –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –Њ–±—Г—Б–ї–Њ–≤–Є–ї–Є –ї–Њ–Љ–∞–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Є—Е –Ї—А–Є–≤—Л—Е –њ–Њ—Б–ї–µ –Љ–∞–Ї—Б–Є–Љ—Г–Љ–∞ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–є –Є –Є—Б–Ї–ї—О—З–Є–ї–Є –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—М –Ї–Њ—А—А–µ–Ї—В–љ–Њ–≥–Њ —А–∞—Б—З–µ—В–∞ –Ї–Њ–љ—Б—В–∞–љ—В—Л —Н–ї–Є–Љ–Є–љ–∞—Ж–Є–Є.
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Ї–Њ—Н–љ–Ј–Є–Љ Q10 –≤ —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є —Д–Њ—А–Љ–µ –Њ–±–ї–∞–і–∞–µ—В –љ–µ—Б–Њ–Љ–љ–µ–љ–љ—Л–Љ–Є –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–∞–Љ–Є: –±–Њ–ї—М—И–µ–є —Б—В–µ–њ–µ–љ—М—О –≤—Б–∞—Б—Л–≤–∞–љ–Є—П, –±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Є–Љ–Є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П–Љ–Є –≤ –њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є –Є –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ – –±–Њ–ї—М—И–µ–є –±–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В—М—О. –≠—В–Њ —Б–Њ–≥–ї–∞—Б—Г–µ—В—Б—П —Б –Є–Љ–µ—О—Й–Є–Љ–Є—Б—П –≤ –ї–Є—В–µ—А–∞—В—Г—А–µ –і–∞–љ–љ—Л–Љ–Є, –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—Й–Є–Љ–Є, —З—В–Њ –њ—А–Є –і–ї–Є—В–µ–ї—М–љ–Њ–Љ –њ—А–Є–µ–Љ–µ —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е —Д–Њ—А–Љ –њ–ї–∞–Ј–Љ–µ–љ–љ—Л–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 –≤ 2–2,5 —А–∞–Ј–∞ –≤—Л—И–µ [4], –∞ –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–∞—П –±–Є–Њ–і–Њ—Б—В—Г–њ–љ–Њ—Б—В—М –≤ 3–6 —А–∞–Ј –±–Њ–ї—М—И–µ, —З–µ–Љ –њ—А–Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–Є –њ–Њ—А–Њ—И–Ї–∞ [6,12]. –§–∞—А–Љ–∞–Ї–Њ–Ї–Є–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–∞ —Б–Њ–ї—О–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є —Д–Њ—А–Љ—Л –Њ–±—Г—Б–ї–Њ–≤–Є–ї–Є –µ–µ –≤—Л—Б–Њ–Ї—Г—О —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –≤ –Ї–∞—З–µ—Б—В–≤–µ –Ї–∞—А–і–Є–Њ–њ—А–Њ—В–µ–Ї—В–Њ—А–∞: —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –њ–µ—А–Њ—А–∞–ї—М–љ–Њ–µ –≤–≤–µ–і–µ–љ–Є–µ –њ—А–Є–≤–Њ–і–Є–ї–Њ –Ї –њ–Њ–≤—Л—И–µ–љ–Є—О –љ–µ —В–Њ–ї—М–Ї–Њ –њ–ї–∞–Ј–Љ–µ–љ–љ—Л—Е —Г—А–Њ–≤–љ–µ–є –Ї–Њ—Н–љ–Ј–Є–Љ–∞ Q10 (–≤ 2,5 —А–∞–Ј–∞), –љ–Њ –Є —Б–Њ–і–µ—А–ґ–∞–љ–Є—П –µ–≥–Њ –≤ –Љ–Є–Њ–Ї–∞—А–і–µ –Ї—А—Л—Б, —З—В–Њ —Г–≤–µ–ї–Є—З–Є–≤–∞–ї–Њ –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М –Ї–ї–µ—В–Њ–Ї –Љ–Є–Њ–Ї–∞—А–і–∞ –≤ —Г—Б–ї–Њ–≤–Є—П—Е –Є—И–µ–Љ–Є–Є –Є –≤ –Є—В–Њ–≥–µ –Њ–≥—А–∞–љ–Є—З–Є–≤–∞–ї–Њ —А–∞–Ј–Љ–µ—А –њ–Њ—Б—В–Є–љ—Д–∞—А–Ї—В–љ–Њ–є –Ј–Њ–љ—Л –љ–µ–Ї—А–Њ–Ј–∞ [2].
–Ы–Є—В–µ—А–∞—В—Г—А–∞
1. –Р—А–Њ–љ–Њ–≤ –Ф.–Ь. // –†—Г—Б—Б–Ї–Є–є –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Є–є –ґ—Г—А–љ–∞–ї. 2004. –Ґ.12. вДЦ 15. –°. 905–909.
2. –Ъ–∞–ї–µ–љ–Є–Ї–Њ–≤–∞ –Х.–Ш., –У–Њ—А–Њ–і–µ—Ж–Ї–∞—П –Х.–Р., –Ъ–Њ–ї–Њ–Ї–Њ–ї—М—З–Є–Ї–Њ–≤–∞ –Х. –Є –і—А. // –С–Є–Њ—Е–Є–Љ–Є—П. 2007. –Ґ. 72. вДЦ 3. –°. 407–415.
3. Bhagavan H.N., Chopra R.K. // Free Radical Res. 2006. V. 40. P. 445–453.
4. Chopra R.K., Godman R., Sinatra S.T., and Bhagavan H.N. // Int. J. Vitam. Nutr. Res. 1998. V. 68. вДЦ 2. P.109–113.
5. Lass A., and Sohal R. // Free Radic. Biol. & Med. 1998. V. 27. P. 220–226.
6. Miles M., Horn P., Miles L. et al. // Nutr. Res. 2002. V. 22. P. 919–929.
7. Sarter B. // J. Cardiovasc. Nurs. 2002. V. 16. P. 9–20.
8. Ullman U., Metzner J., Schulz C. et al. // Journal of Medical Food. 2005. V.8. вДЦ 3. P. 397–399.
9. Weant K.A., and Smith K.M. // Ann. Pharmacother. 2005. V. 39. P. 1522–1526.
10. Weiss M., Mortensen S.A., Rassing M.R. et al. // Mol. Aspects Med. 1994. V. 15. P. s273–s280.
11. Yuzuriha T., Takada M., Katayama K. // Biochim. Biophys. Acta. 1983. V. 759. P. 286–291.
12. Zaghloul A., Gurley B., Khan M. et al. // Drug. Dev. Ind. Pharm. 2002. V. 28. вДЦ 10. P. 1195–1200.
13. Zhang Y., Aberg F., Appelkvist E–L. et al. // J. Nutr. 1995. V. 125. P. 446–453.



