




–Т —Н—В–Њ–є —Б—В–∞—В—М–µ –Љ—Л –Њ–њ–Є—Б—Л–≤–∞–µ–Љ —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л–µ –њ–Њ–і—Е–Њ–і—Л, –њ—А–µ–і–ї–Њ–ґ–µ–љ–љ—Л–µ –і–ї—П –ї–µ—З–µ–љ–Є—П —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –±–Њ–ї–µ–Ј–љ–Є –њ–Њ—З–µ–Ї (–•–С–Я). –Я–Њ—Б–Ї–Њ–ї—М–Ї—Г –ї–Є—И—М –љ–µ–±–Њ–ї—М—И–Њ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є [6,7] –њ—А–Њ–≤–Њ–і–Є–ї–Њ—Б—М –њ–Њ –њ—А–Њ—В–Њ–Ї–Њ–ї–∞–Љ, –њ–Њ–Ј–≤–Њ–ї—П—О—Й–Є–Љ –Љ–Њ–љ–Є—В–Њ—А–Є—А–Њ–≤–∞—В—М –њ–Њ—З–µ—З–љ—Г—О —Д—Г–љ–Ї—Ж–Є—О (–њ—А–Њ—В–Њ–Ї–Њ–ї—Л, –≤ –Ї–Њ—В–Њ—А—Л—Е –ї–µ—З–µ–љ–Є–µ –љ–∞—З–Є–љ–∞–ї–Њ—Б—М –њ–Њ—Б–ї–µ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П —А–µ–љ–∞–ї—М–љ—Л—Е –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–є), –Љ—Л –Њ–±—Б—Г–ґ–і–∞–µ–Љ –љ–∞–Є–±–Њ–ї–µ–µ –≤–∞–ґ–љ—Л–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –≤ –Ї–Њ—В–Њ—А—Л—Е –Њ–њ—А–µ–і–µ–ї–µ–љ—Л –Љ–Є—И–µ–љ–Є –і–ї—П –њ—А–µ–і—Г–њ—А–µ–ґ–і–µ–љ–Є—П —А–∞–Ј–≤–Є—В–Є—П –њ–Њ—З–µ—З–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є (–≤ —Н—В–Є—Е –њ—А–Њ—В–Њ–Ї–Њ–ї–∞—Е –ї–µ—З–µ–љ–Є–µ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–ї–Њ—Б—М –Є–љ–і—Г–Ї—Ж–Є–µ–є –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞).
–Я—А–Њ—В–Є–≤–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –∞–≥–µ–љ—В—Л
–°—З–Є—В–∞–µ—В—Б—П, —З—В–Њ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ –Є–≥—А–∞–µ—В –≤–∞–ґ–љ—Г—О —А–Њ–ї—М –≤ –Ј–∞–њ—Г—Б–Ї–µ –±–Њ–ї–µ–Ј–љ–µ–є –њ–Њ—З–µ–Ї. –Я—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ —Б—В—А–µ–њ—В–Њ–Љ–Є—Ж–Є–љ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є –љ–∞ –Љ—Л—И–∞—Е –±—Л–ї–Њ –Ј–∞–Љ–µ–і–ї–µ–љ–Њ –Є–Ј––Ј–∞ –љ–µ–і–Њ—Б—В–∞—В–Ї–∞ —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є —Е–µ–Љ–Њ–∞—В—В—А–∞–Ї—В–∞–љ—В–љ—Л—Е –Љ–Њ–љ–Њ—Ж–Є—В–Њ–≤ –њ—А–Њ—В–µ–Є–љ–∞–1 [8]. –≠—Д—Д–µ–Ї—В—Л –њ—А–Њ—В–Є–≤–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–є —В–µ—А–∞–њ–Є–Є –±—Л–ї–Є –Є–Ј—Г—З–µ–љ—Л –љ–∞ –Љ–Њ–і–µ–ї—П—Е —Б–µ—А–њ–Њ–≤–Є–і–љ–Њ–≥–Њ –≥–ї–Њ–Љ–µ—А—Г–ї–Њ–љ–µ—Д—А–Є—В–∞ –љ–∞ –Ї—А—Л—Б–∞—Е. –Ъ–Њ–≥–і–∞ –ї–µ—З–µ–љ–Є–µ –љ–∞—З–Є–љ–∞–ї–Њ—Б—М –њ—А–Є –і–µ–±—О—В–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, —В–µ—З–µ–љ–Є–µ –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ —Б–Ї–ї–µ—А–Њ–Ј–∞ –±—Л–ї–Њ –Ј–∞–Љ–µ—В–љ–Њ –ї—Г—З—И–µ, –∞ –њ—А–Њ—В–µ–Є–љ—Г—А–Є—П –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ —Б–љ–Є–ґ–µ–љ–∞. –Ю–і–љ–∞–Ї–Њ —Г–ї—Г—З—И–µ–љ–Є—П –љ–µ –љ–∞—Б—В—Г–њ–∞–ї–Њ –њ—А–Є –љ–∞—З–∞–ї–µ –ї–µ—З–µ–љ–Є—П –≤ –њ–Њ–Ј–і–љ—О—О —Д–∞–Ј—Г, –њ–Њ—Б–ї–µ –њ–Њ—П–≤–ї–µ–љ–Є—П –≥–ї–Њ–Љ–µ—А—Г–ї—П—А–љ–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є [9]. –†–∞–њ–∞–Љ–Є—Ж–Є–љ, –Є—Б–њ–Њ–ї—М–Ј—Г–µ–Љ—Л–є –≤ –Њ—Б–љ–Њ–≤–љ–Њ–Љ –Ї–∞–Ї –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Њ—А, —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞–ї —Г–Љ–µ–љ—М—И–µ–љ–Є—О —В—Г–±—Г–ї—П—А–љ–Њ–є –і–Є–ї–∞—В–∞—Ж–Є–Є, –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –Њ–±—К–µ–Љ–∞ –Є –Њ—В–ї–Њ–ґ–µ–љ–Є—О –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ –њ—А–Є –Њ–і–љ–Њ—Б—В–Њ—А–Њ–љ–љ–µ–є –Љ–Њ—З–µ—В–Њ—З–љ–Є–Ї–Њ–≤–Њ–є –Њ–±—Б—В—А—Г–Ї—Ж–Є–Є (–Ю–Ь–Ю) [10]. –Я–Њ–і–Њ–±–љ—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л —В–∞–Ї–ґ–µ –љ–∞–±–ї—О–і–∞–ї–Є—Б—М –њ—А–Є –≤–≤–µ–і–µ–љ–Є–Є –Є–љ–≥–Є–±–Є—В–Њ—А–Њ–≤ –љ—Г–Ї–ї–µ–∞—А–љ–Њ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ (NF) – kappa–Т, –Є–ї–Є –Ї—Г—А–Ї—Г–Љ–Є–љ–∞, –љ–∞ —В–µ—Е –ґ–µ —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –Љ–Њ–і–µ–ї—П—Е [11]. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –њ–Њ–і–∞–≤–ї–µ–љ–Є–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ –љ–∞ —А–∞–љ–љ–Є—Е —Б—В–∞–і–Є—П—Е —А–∞–Ј–≤–Є—В–Є—П –•–С–Я, –љ–Њ –µ–≥–Њ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –і–Њ–≤–Њ–ї—М–љ–Њ –Њ–≥—А–∞–љ–Є—З–µ–љ–∞.
–Р–љ—В–∞–≥–Њ–љ–Є—Б—В—Л —В—А–∞–љ—Б—Д–Њ—А–Љ–Є—А—Г—О—Й–µ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ —А–Њ—Б—В–∞–b
–Я–Њ–ї–∞–≥–∞—О—В, —З—В–Њ —В—А–∞–љ—Б—Д–Њ—А–Љ–Є—А—Г—О—Й–Є–є —Д–∞–Ї—В–Њ—А —А–Њ—Б—В–∞–b (transforming growth factor–beta, TGFb) —П–≤–ї—П–µ—В—Б—П –≥–ї–∞–≤–љ—Л–Љ –∞–Ї—В–Є–≤–∞—В–Њ—А–Њ–Љ —Б–Є–љ—В–µ–Ј–∞ —Н–Ї—Б—В—А–∞—Ж–µ–ї—О–ї—П—А–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞. –Я—А–µ–і–ї–∞–≥–∞–µ–Љ—Л–µ —А–∞–љ–µ–µ –Љ–µ—В–Њ–і—Л (–Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –і–µ–Ї–∞—А–Є–љ–∞ – –Њ—З–Є—Б—В–Є—В–µ–ї—П TGFb –Є–ї–Є —А–∞—Б—В–≤–Њ—А–Є–Љ—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤) –µ–≥–Њ –Є–Ј—Г—З–µ–љ–Є—П –±—Л–ї–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –Њ–≥—А–∞–љ–Є—З–µ–љ—Л –≤ –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Њ–Љ –њ—А–Є–Љ–µ–љ–µ–љ–Є–Є –Є –љ–µ –њ–Њ–Ј–≤–Њ–ї—П–ї–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞—В—М –µ–≥–Њ –љ–∞ –ї—О–і—П—Е (–њ–Њ–≤—Л—И–∞–ї–∞—Б—М –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –і–µ–Ї–∞—А–Є–љ–∞, —З—В–Њ –њ—А–Є–≤–Њ–і–Є–ї–Њ –Ї –љ–µ–і–Њ—Б—В–∞—В–Ї—Г –∞–љ—В–Є–TGFb —Б–њ–µ—Ж–Є—Д–Є—З–љ–Њ—Б—В–Є; –Ї —В–Њ–Љ—Г –ґ–µ —Б—В–Њ–Є–Љ–Њ—Б—В—М –Є –Љ–µ—В–Њ–і –і–Њ—Б—В–∞–≤–Ї–Є —А–∞—Б—В–≤–Њ—А–Є–Љ—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –і–µ–ї–∞–ї–Є —Б—В–Њ–ї—М –і–ї–Є—В–µ–ї—М–љ—Г—О —В–µ—А–∞–њ–Є—О –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –љ–µ–≤–Њ–Ј–Љ–Њ–ґ–љ–Њ–є).
–Я–Њ–Ј–ґ–µ –±—Л–ї–Њ –њ—А–µ–і–ї–Њ–ґ–µ–љ–Њ –±–ї–Њ–Ї–Є—А–Њ–≤–∞—В—М —Д–Є–±—А–Њ–≥–µ–љ–љ—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М TGFb —Б –њ–Њ–Љ–Њ—Й—М—О –∞–≥–µ–љ—В–Њ–≤, –Є–љ–≥–Є–±–Є—А—Г—О—Й–Є—Е TGFb –Є–ї–Є —П–≤–ї—П—О—Й–Є—Е—Б—П –µ–≥–Њ –∞–љ—В–∞–≥–Њ–љ–Є—Б—В–∞–Љ–Є. –Ш—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–ї–Є –Ї–Њ—Б—В–љ—Л–є –Љ–Њ—А—Д–Њ–≥–µ–љ–љ—Л–є –њ—А–Њ—В–µ–Є–љ 7 (bone morphogenic protein 7 – BMP7) –Є —Д–∞–Ї—В–Њ—А —А–Њ—Б—В–∞ –≥–µ–њ–∞—В–Њ—Ж–Є—В–Њ–≤ (hepatocyte growth factor, HGF). –Т –і–≤—Г—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –њ—А–Є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є –±—Л–ї –Є–Ј—Г—З–µ–љ —Н—Д—Д–µ–Ї—В BMP7. –Т –њ–µ—А–≤–Њ–Љ —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–µ –≤ —В–µ—З–µ–љ–Є–µ 1 –≥–Њ–і–∞ –Є–Ј—Г—З–∞–ї–Є—Б—М —В—А–∞–љ—Б–≥–µ–љ–љ—Л–µ –Љ—Л—И–Є, —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г—О—Й–Є–µ —Д–Њ—Б—Д–Њ–µ–љ–Њ–ї–њ–Є—А—Г–≤–∞—В –Ї–∞—А–±–Њ–Ї—Б–Є–Ї–Є–љ–∞–Ј—Г, –Ї–Њ—В–Њ—А–∞—П –∞–Ї—В–Є–≤–Є–Ј–Є—А—Г–µ—В —В—А–∞–љ—Б–≥–µ–љ BMP7. –Ґ—А–∞–љ—Б–≥–µ–љ–љ–∞—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—П BMP7 –≤ –њ–Њ–і–Њ—Ж–Є—В–∞—Е –Ї–ї—Г–±–Њ—З–Ї–∞ –Є –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–∞—Е –њ—А–µ–і—Г–њ—А–µ–ґ–і–∞–ї–∞ —Г–Љ–µ–љ—М—И–µ–љ–Є–µ —З–Є—Б–ї–∞ –њ–Њ–і–Њ—Ж–Є—В–Њ–≤. –≠–Ї—Б–њ—А–µ—Б—Б–Є—П BMP7 —В–∞–Ї–ґ–µ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–ї–∞—Б—М —Г–Љ–µ–љ—М—И–µ–љ–Є–µ–Љ –≥–ї–Њ–Љ–µ—А—Г–ї—П—А–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞, –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ–Љ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –Ї–Њ–ї–ї–∞–≥–µ–љ–∞, –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ I –Є —Н–Ї—Б–њ—А–µ—Б—Б–Є–µ–є —Д–Є–±—А–Њ–љ–µ–Ї—В–Є–љ–∞ [12]. –Т –і—А—Г–≥–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –≤–≤–µ–і–µ–љ–Є–µ —Н–Ї–Ј–Њ–≥–µ–љ–љ–Њ–≥–Њ BMP7 –њ–Њ–і–∞–≤–ї—П–ї–Њ —А–∞–Ј–≤–Є—В–Є–µ —А–µ–љ–∞–ї—М–љ—Л—Е –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–є, —В–∞–Ї–Є—Е –Ї–∞–Ї –≥–ї–Њ–Љ–µ—А—Г–ї—П—А–љ–∞—П –≥–Є–њ–µ—А—В—А–Њ—Д–Є—П, –і–Є—Д—Д—Г–Ј–љ—Л–є –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј, —В—Г–±—Г–ї—П—А–љ–∞—П –∞—В—А–Њ—Д–Є—П –Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј [13].
–Э–µ —В–∞–Ї –і–∞–≤–љ–Њ –±—Л–ї –Њ–њ–Є—Б–∞–љ –њ–Њ—В–µ–љ—Ж–Є–∞–ї HGF, –Ї–∞–Ї –∞–љ—В–Є—Д–Є–±—А–Њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –∞–≥–µ–љ—В–∞ [14]. –У–µ–љ–љ–∞—П —В–µ—А–∞–њ–Є—П —З–µ–ї–Њ–≤–µ—З–µ—Б–Ї–Є–Љ HGF (–≤–љ—Г—В—А–Є–Љ—Л—И–µ—З–љ—Л–є –≥–µ–љ–љ—Л–є —Н–ї–µ–Ї—В—А–Њ—В—А–∞–љ—Б—Д–µ—А) —Б–љ–Є–Ј–Є–ї–∞ —Г—А–Њ–≤–µ–љ—М –њ–Њ—З–µ—З–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –Є —Б–Љ–µ—А—В–љ–Њ—Б—В–Є, —З–∞—Б—В–Њ—В—Г –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л—Е —В—Г–±—Г–ї—П—А–љ—Л—Е –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–є, —Б—В–Є–Љ—Г–ї–Є—А–Њ–≤–∞–ї–∞ –Ї–ї–µ—В–Њ—З–љ—Г—О —А–µ–≥–µ–љ–µ—А–∞—Ж–Є—О, —Б–љ–Є–Ј–Є–ї–∞ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ, –∞–Ї—В–Є–≤–∞—Ж–Є—О NF–kappaB, —Б–Њ–Ї—А–∞—В–Є–ї–∞ —Н–Ї—Б–њ—А–µ—Б—Б–Є—О –Љ–∞—А–Ї–µ—А–Њ–≤ —Д–Є–±—А–Њ–Ј–∞ –Є –њ—А–µ–і—Г–њ—А–µ–і–Є–ї–∞ —А–∞–Ј–≤–Є—В–Є–µ –њ–Њ–Ј–і–љ–µ–≥–Њ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ –Є –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –љ–∞ –Љ–Њ–і–µ–ї—П—Е —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –∞–ї–ї–Њ—В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—В–љ–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є. –Ы–µ—З–µ–љ–Є–µ —Б –њ–Њ–Љ–Њ—Й—М—О HGF –Њ–Ї–∞–Ј–∞–ї–Њ—Б—М –≥–Њ—А–∞–Ј–і–Њ –±–Њ–ї–µ–µ —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–Љ –≤ –Ј–∞–і–µ—А–ґ–Ї–µ —А–∞–Ј–≤–Є—В–Є—П –њ–Њ—З–µ—З–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ [15]. –Ф—А—Г–≥–Є–µ —Г—З–µ–љ—Л–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–ї–Є –Є–љ–≥–Є–±–Є—В–Њ—А ALK5––Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ–љ–Њ–≥–Њ Smad3, –њ—А–µ–і—Г–њ—А–µ–ґ–і–∞—О—Й–Є–є –Ї–∞—Б–Ї–∞–і –і–µ–є—Б—В–≤–Є–є TGFb (IN–1130) –њ—А–Є –Ю–Ь–Ю –љ–∞ –Ї—А—Л—Б–∞—Е. –•–∞—А–∞–Ї—В–µ—А–љ—Л–µ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, –њ—А–Є—Б—Г—Й–Є–µ –Ю–Ь–Ю, —В–∞–Ї–Є–µ –Ї–∞–Ї –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј, –∞—В—А–Њ—Д–Є—П –Є —А–∞—Б—И–Є—А–µ–љ–Є–µ –Ї–∞–љ–∞–ї—М—Ж–µ–≤, –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–∞—П –Ї–ї–µ—В–Њ—З–љ–∞—П –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є—П –Є —Д–Є–±—А–Њ–±–ї–∞—Б—В–љ–∞—П –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є—П, –±—Л–ї–Є –њ—А–µ–Њ—В–≤—А–∞—Й–µ–љ—Л –њ—А–Є —В–µ—А–∞–њ–Є–Є IN–1130. –≠—В–Њ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–ї–Њ—Б—М —Б–љ–Є–ґ–µ–љ–Є–µ–Љ —Г—А–Њ–≤–љ—П TGFb, –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ I —В–Є–њ–∞, pSmad2 –Є —Д–Є–±—А–Њ–љ–µ–Ї—В–Є–љ–∞ [16].
–Ґ—А–∞–љ–Є–ї–∞—Б—В —П–≤–ї—П–µ—В—Б—П –∞–љ—В–Є—Д–Є–±—А–Њ—В–Є—З–µ—Б–Ї–Є–Љ –∞–≥–µ–љ—В–Њ–Љ, –Є–љ–≥–Є–±–Є—А—Г—О—Й–Є–Љ TGFb; –≤—Л—Б–≤–Њ–±–Њ–ґ–і–∞–µ—В—Б—П –Є–Ј —А–∞–Ј–ї–Є—З–љ—Л—Е –Ї–ї–µ—В–Њ–Ї, –≤–Ї–ї—О—З–∞—П —Д–Є–±—А–Њ–±–ї–∞—Б—В—Л –Є –Љ–∞–Ї—А–Њ—Д–∞–≥–Є. –Э–∞ –Љ–Њ–і–µ–ї—П—Е —Б—Г–±—В–Њ—В–∞–ї—М–љ–Њ–є –љ–µ—Д—А—Н–Ї—В–Њ–Љ–Є–Є –≤–≤–µ–і–µ–љ–Є–µ —В—А–∞–љ–Є–ї–∞—Б—В–∞ –Њ—Б–ї–∞–±–ї—П–ї–Њ —Б—В–µ–њ–µ–љ—М —В—А–∞–≤–Љ–∞—В–Є–Ј–∞—Ж–Є–Є –њ–Њ—З–Ї–Є. –Ш–љ—В–µ—А–µ—Б–љ–Њ, —З—В–Њ –Њ–і–љ–Њ–Љ–Њ–Љ–µ–љ—В–љ–Њ–µ –≤–≤–µ–і–µ–љ–Є–µ –Є–љ–≥–Є–±–Є—В–Њ—А–∞ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–њ—А–µ–≤—А–∞—Й–∞—О—Й–µ–≥–Њ —Д–µ—А–Љ–µ–љ—В–∞ (–Є–Р–Я–§) –≤ –і–∞–ї—М–љ–µ–є—И–µ–Љ —Г–≤–µ–ї–Є—З–Є–≤–∞–ї–Њ –Ї–ї–Є—А–µ–љ—Б –Ї—А–µ–∞—В–Є–љ–Є–љ–∞, —Б–љ–Є–ґ–∞–ї–Њ –њ—А–Њ—В–µ–Є–љ—Г—А–Є—О –Є –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј, —З—В–Њ —Г–Ї–∞–Ј—Л–≤–∞–µ—В –љ–∞ –њ—А–Є–µ–Љ–ї–µ–Љ–Њ—Б—В—М —В–∞–Ї–Њ–є –і–≤–Њ–є–љ–Њ–є —В–µ—А–∞–њ–Є–Є –њ—А–Є –њ–Њ—З–µ—З–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е [17]. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, —Г –љ–µ–±–Њ–ї—М—И–Њ–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б —А–∞–љ–љ–Є–Љ–Є —Б—В–∞–і–Є—П–Љ–Є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є –ї–µ—З–µ–љ–Є–µ —В—А–∞–љ–Є–ї–∞—Б—В–Њ–Љ –≤ —В–µ—З–µ–љ–Є–µ 1 –≥–Њ–і–∞ –љ–∞–±–ї—О–і–∞–ї–∞—Б—М —В–µ–љ–і–µ–љ—Ж–Є—П –Ї —Б–љ–Є–ґ–µ–љ–Є—О —Н–Ї—Б–Ї—А–µ—Ж–Є–Є –Њ–±–Њ–Є—Е —В–Є–њ–Њ–≤ –њ–Њ—З–µ—З–љ–Њ–≥–Њ –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ IV –Є –∞–ї—М–±—Г–Љ–Є–љ–∞ [18]. –Я—А–µ–ґ–і–µ —З–µ–Љ –і–µ–ї–∞—В—М –≤—Л–≤–Њ–і—Л –Њ–± —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є —В—А–∞–љ–Є–ї–∞—Б—В–∞ –њ—А–Є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ—А–Њ–≤–µ–і–µ–љ–Є–µ –Ї—А—Г–њ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є —Б –±–Њ–ї–µ–µ –і–ї–Є—В–µ–ї—М–љ—Л–Љ –њ–µ—А–Є–Њ–і–Њ–Љ –љ–∞–±–ї—О–і–µ–љ–Є—П. –Я—А–Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–Є –љ–∞ –Љ–Њ–і–µ–ї—П—Е –Ю–Ь–Ю –і–≤–Њ–є–љ–Њ–є —В–µ—А–∞–њ–Є–Є, —Б–Њ—Б—В–Њ—П—Й–µ–є –Є–Ј –Љ–Њ–љ–Њ–Ї–ї–Њ–љ–∞–ї—М–љ—Л—Е –∞–љ—В–Є—В–µ–ї –њ—А–Њ—В–Є–≤ TGFb (1D11) –Є –Є–Р–Я–§, –±—Л–ї–Є –њ–Њ–ї—Г—З–µ–љ—Л –њ—А–Њ—В–Є–≤–Њ—А–µ—З–Є–≤—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л. –Т–≤–µ–і–µ–љ–Є–µ –Њ–±–Њ–Є—Е 1D11 –Є–ї–Є –Є–Р–Я–§ –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ–Њ —Г–Љ–µ–љ—М—И–∞–ї–Њ —Д–Є–±—А–Њ–Ј–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–ї—М–љ—Г—О –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є—О, –∞–њ–Њ–њ—В–Њ–Ј –Є —Н–Ї—Б–њ—А–µ—Б—Б–Є—О TGFb. –Ъ–Њ–Љ–±–Є–љ–Є—А–Њ–≤–∞–љ–љ–∞—П —В–µ—А–∞–њ–Є—П –љ–µ –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–ї–∞ —Г—Б–Є–ї–µ–љ–Є–µ —Н—Д—Д–µ–Ї—В–∞, –њ–Њ–Ї–∞–Ј—Л–≤–∞—П —В–µ–Љ —Б–∞–Љ—Л–Љ, —З—В–Њ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ II –Є TGFb —П–≤–ї—П—О—В—Б—П –Ј–≤–µ–љ—М—П–Љ–Є –Њ–і–љ–Њ–є —Ж–µ–њ–Є [19]. –Я–Њ–і–∞–≤–ї–µ–љ–Є–µ —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є TGFb, –њ—А–Є–Љ–µ–љ—П–µ–Љ–Њ–є –њ—А–Є siRNA –Љ–µ—В–Њ–і–Є–Ї–µ, –њ—А–µ–і—Г–њ—А–µ–ґ–і–∞–ї–Њ —А–∞–љ–љ—О—О —Н–Ї—Б–њ—А–µ—Б—Б–Є—О –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ I —В–Є–њ–∞ –Є –Ј–∞–Љ–µ–і–ї—П–ї–Њ —А–∞–Ј–≤–Є—В–Є–µ —Д–Є–±—А–Њ–Ј–љ—Л—Е –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–є –љ–∞ –Љ–Њ–і–µ–ї—П—Е –Ю–Ь–Ю [20].
–Ш–љ–≥–Є–±–Є—В–Њ—А—Л —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –Ї —Д–∞–Ї—В–Њ—А—Г —А–Њ—Б—В–∞ —В–Є—А–Њ–Ј–Є–љ–Ї–Є–љ–∞–Ј—Л
–Я–Њ –љ–µ–і–∞–≤–љ–Є–Љ –љ–∞–±–ї—О–і–µ–љ–Є—П–Љ, —А–µ—Ж–µ–њ—В–Њ—А—Л –Ї —В–Є—А–∞–Ј–Є–љ–Ї–Є–љ–∞–Ј–љ–Њ–Љ—Г —Д–∞–Ї—В–Њ—А—Г —А–Њ—Б—В–∞ –Љ–Њ–≥—Г—В –±—Л—В—М –∞–Ї—В–Є–≤–Є—А–Њ–≤–∞–љ—Л —Б –њ–Њ–Љ–Њ—Й—М—О –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–∞ –Є–ї–Є —Н–љ–і–Њ—В–µ–ї–Є–љ–∞. –≠—В–Њ –Њ—В–Ї—А—Л–≤–∞–µ—В –љ–Њ–≤—Л–µ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є, —В–∞–Ї –Ї–∞–Ї –±–ї–Њ–Ї–∞—В–Њ—А—Л —Н—В–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Њ—Б—В–∞ –Љ–Њ–≥—Г—В –±—Л—В—М –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ—Л —Б–Њ –≤—А–µ–Љ–µ–љ–µ–Љ –≤ –ї–µ—З–µ–љ–Є–Є –њ–Њ—З–µ—З–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞. –Т—Л—П–≤–ї–µ–љ–Њ, —З—В–Њ –±–ї–Њ–Ї–∞–і–∞ EGF–—А–µ—Ж–µ–њ—В–Њ—А–∞ –њ—А–µ–і—Г–њ—А–µ–ґ–і–∞–µ—В —А–∞–Ј–≤–Є—В–Є–µ —А–µ–љ–∞–ї—М–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—Й–µ–≥–Њ –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–≤–љ—Г—О –љ–µ—Д—А–Њ–њ–∞—В–Є—О [21]. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –Ї —В—А–Њ–Љ–±–Њ—Ж–Є—В–∞—А–љ–Њ–Љ—Г —Д–∞–Ї—В–Њ—А—Г —А–Њ—Б—В–∞ (platelet–derived growth factor, PDGF), –њ—А–Њ–≤–µ–і–µ–љ–љ—Л–µ –≤ –њ—А–Њ—И–ї–Њ–Љ –≥–Њ–і—Г, –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ –Є–Љ–∞—В–Є–љ–Є–± (–Є–љ–≥–Є–±–Є—В–Њ—А PDGF–Rbeta) —Г–ї—Г—З—И–∞–ї —Д—Г–љ–Ї—Ж–Є—О –Є —Б–љ–Є–ґ–∞–ї –њ–Њ—З–µ—З–љ—Л–µ –Љ–Є–Ї—А–Њ–≤–∞—Б–Ї—Г–ї—П—А–љ—Л–µ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –љ–µ–Ј–∞–≤–Є—Б–Є–Љ–Њ –Њ—В —Г—А–Њ–≤–љ—П –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П —Г —В—А–∞–љ—Б–≥–µ–љ–љ—Л—Е –Ї—А—Л—Б —Б –њ–Њ–≤—Л—И–µ–љ–љ–Њ–є –≤—Л—А–∞–±–Њ—В–Ї–Њ–є —А–µ–љ–Є–љ–∞ [22]. –†–∞–љ–љ–µ–µ –Є–ї–Є –њ–Њ–Ј–і–љ–µ–µ –≤–≤–µ–і–µ–љ–Є–µ PDGF–D––љ–µ–є—В—А–∞–ї–Є–Ј—Г—О—Й–Є—Е –∞–љ—В–Є—В–µ–ї —Г–ї—Г—З—И–∞–ї–Њ —П–≤–ї–µ–љ–Є—П —Д–Њ–Ї–∞–ї—М–љ–Њ–≥–Њ —Б–µ–≥–Љ–µ–љ—В–∞—А–љ–Њ–≥–Њ –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞, –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–є –њ–Њ–і–Њ—Ж–Є—В–Њ–≤ –Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є—П –Ї–∞–љ–∞–ї—М—Ж–µ–≤, –∞ —В–∞–Ї–ґ–µ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –њ–Њ—З–µ—З–љ–Њ–≥–Њ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞ (–Ї–Њ–ї–ї–∞–≥–µ–љ III —В–Є–њ–∞ –Є —Д–Є–±—А–Њ–љ–µ–Ї—В–Є–љ), —Г–Љ–µ–љ—М—И–∞–ї–Њ –Ї–Њ—А—В–Є–Ї–∞–ї—М–љ—Г—О –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є—О –Љ–∞–Ї—А–Њ—Д–∞–≥–∞–Љ–Є –Є –њ—А–Њ—В–µ–Є–љ—Г—А–Є—О –љ–∞ –Љ–Њ–і–µ–ї—П—Е –Ї—А—Л—Б –≥–ї–Њ–Љ–µ—А—Г–ї–Њ–љ–µ—Д—А–Є—В–∞ (–Њ–і–љ–Њ—Б—В–Њ—А–Њ–љ–љ—П—П –љ–µ—Д—А—Н–Ї—В–Њ–Љ–Є—П –њ–ї—О—Б –∞–љ—В–Є–Thy–1.1 –∞–љ—В–Є—В–µ–ї–∞). –Ф–∞–љ–љ—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л –≥–Њ–≤–Њ—А—П—В –Њ –≤–∞–ґ–љ–Њ–є —А–Њ–ї–Є PDGF–D –≤ –њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ —Б–Љ–Њ—А—Й–Є–≤–∞–љ–Є—П –њ–Њ—З–Ї–Є –Є –≤—Л–і–µ–ї—П—О—В –љ–Њ–≤—Л–є —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Є–є –њ–Њ–і—Е–Њ–і –Ї –ї–µ—З–µ–љ–Є—О –Љ–µ–Ј–∞–љ–≥–Є–Њ–њ—А–Њ–ї–Є—Д–µ—А–∞—В–Є–≤–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –њ–Њ—З–µ–Ї [23,24].
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –∞–љ—В–∞–≥–Њ–љ–Є—Б—В–Њ–≤ —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –Ї —Д–∞–Ї—В–Њ—А–∞–Љ —А–Њ—Б—В–∞ –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є –Є–љ—В–µ—А–µ—Б–љ—Л–є –њ–Њ–і—Е–Њ–і –Ї –∞–љ—В–Є—Д–Є–±—А–Њ—В–Є—З–µ—Б–Ї–Њ–є —В–µ—А–∞–њ–Є–Є. –Ю–і–љ–∞–Ї–Њ –Њ–њ—Л—В —Б –і–ї–Є—В–µ–ї—М–љ–Њ–є –±–ї–Њ–Ї–∞–і–Њ–є —Д–∞–Ї—В–Њ—А–∞ —А–Њ—Б—В–∞, –њ—А–Њ–≤–µ–і–µ–љ–љ—Л–є –љ–∞ –ї—О–і—П—Е, –≤—Л—П–≤–Є–ї –љ–µ–ґ–µ–ї–∞—В–µ–ї—М–љ—Л–µ —Н—Д—Д–µ–Ї—В—Л. –Ґ–∞–Ї –Ї–∞–Ї —А–µ–љ–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј –љ—Г–ґ–і–∞–µ—В—Б—П –≤ –і–ї–Є—В–µ–ї—М–љ–Њ–є —В–µ—А–∞–њ–Є–Є, –љ–∞–Є–±–Њ–ї–µ–µ –њ—А–∞–≤–Є–ї—М–љ–Њ–є —П–≤–ї—П–µ—В—Б—П –Ї–Њ–Љ–±–Є–љ–∞—Ж–Є—П –Ї–Њ—А–Њ—В–Ї–Є—Е –њ–µ—А–Є–Њ–і–Њ–≤ –Є–љ–≥–Є–±–Є—А–Њ–≤–∞–љ–Є—П —Д–∞–Ї—В–Њ—А–∞ —А–Њ—Б—В–∞ —Б —В—А–∞–і–Є—Ж–Є–Њ–љ–љ–Њ–є –і–ї–Є—В–µ–ї—М–љ–Њ–є –љ–µ—Д—А–Њ–њ—А–Њ—В–µ–Ї—В–Є–≤–љ–Њ–є —В–µ—А–∞–њ–Є–µ–є.
–Ш–љ–≥–Є–±–Є—В–Њ—А—Л –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ—Л—Е
—Б–Є–≥–љ–∞–ї—М–љ—Л—Е –њ—Г—В–µ–є
–Т–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є–µ –љ–µ—В–Њ–Ї—Б–Є—З–љ—Л—Е –Є–љ–≥–Є–±–Є—В–Њ—А–Њ–≤ —А–∞–Ј–ї–Є—З–љ—Л—Е —Б–Є–≥–љ–∞–ї—М–љ—Л—Е –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є –њ–Њ—В—А–µ–±–Њ–≤–∞–ї–Њ –њ—А–Њ–≤–µ–і–µ–љ–Є—П –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –Є—Е –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –љ–∞ –њ–Њ—З–µ—З–љ—Г—О —Д—Г–љ–Ї—Ж–Є—О. –Ґ–∞–Ї, –±—Л–ї–Њ –Є–Ј—Г—З–µ–љ–Њ –і–µ–є—Б—В–≤–Є–µ —А38 –Љ–Є—В–Њ–≥–µ–љ––∞–Ї—В–Є–≤–Є—А–Њ–≤–∞–љ–љ–Њ–є –њ—А–Њ—В–µ–Є–љ–Ї–Є–љ–∞–Ј—Л (—А38–Ь–Р–†–Ъ) –њ—А–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –∞–ї–ї–Њ—В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—В–љ–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є, –≤—Л–Ј–≤–∞–љ–љ–Њ–є –≤ —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–µ –љ–∞ –Ї—А—Л—Б–∞—Е. –Т –і–Њ–±–∞–≤–ї–µ–љ–Є–µ –Ї —Б—В–∞–љ–і–∞—А—В–љ–Њ–є —В–µ—А–∞–њ–Є–Є —Ж–Є–Ї–ї–Њ—Б–њ–Њ—А–Є–љ–Њ–Љ –≤–≤–Њ–і–Є–ї—Б—П —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–є –Є–љ–≥–Є–±–Є—В–Њ—А —А38–Ь–Р–†–Ъ. –Ф–∞–љ–љ–∞—П —В–µ—А–∞–њ–Є—П —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞–ї–∞ —Б–Њ—Е—А–∞–љ–µ–љ–Є—О —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї (—Б–љ–Є–ґ–µ–љ–Є–µ–Љ –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞, –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –Њ–±–ї–Є—В–µ—А–∞—Ж–Є–Є, –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ –Є —В—Г–±—Г–ї—П—А–љ–Њ–є –∞—В—А–Њ—Д–Є–Є), –∞ —В–∞–Ї–ґ–µ —Г–≤–µ–ї–Є—З–Є–ї–∞ –њ—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ—Б—В—М –ґ–Є–Ј–љ–Є. –Я—А–µ–і–њ–Њ–ї–∞–≥–∞–µ—В—Б—П, —З—В–Њ –±–ї–Њ–Ї–∞–і–∞ —А38–Ь–Р–†–Ъ —Д–Њ—Б—Д–Њ—А–Є–ї—П—Ж–Є–Є –Љ–Њ–ґ–µ—В –Њ–±–µ—Б–њ–µ—З–Є–≤–∞—В—М –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л–є —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Є–є —Н—Д—Д–µ–Ї—В –≤ –±–Њ—А—М–±–µ —Б —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –∞–ї–ї–Њ—В—А–∞–љ—Б–њ–ї–∞–љ—В–∞—В–љ–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–µ–є [25]. –Ю–і–љ–∞–Ї–Њ –і–∞–љ–љ–∞—П —В–∞–Ї—В–Є–Ї–∞ –љ–∞–Є–±–Њ–ї–µ–µ —Н—Д—Д–µ–Ї—В–Є–≤–љ–∞ –≤ –Њ—В–љ–Њ—И–µ–љ–Є–Є –Ї–∞—А–і–Є–Њ–њ—А–Њ—В–µ–Ї—Ж–Є–Є, —В–Њ–≥–і–∞ –Ї–∞–Ї –љ–µ—Д—А–Њ–њ—А–Њ—В–µ–Ї—В–Є–≤–љ—Л–є —Н—Д—Д–µ–Ї—В –љ–µ–Ј–љ–∞—З–Є—В–µ–ї–µ–љ.
–Я—А–Њ—В–µ–Є–љ–Ї–Є–љ–∞–Ј–∞ –° (–†–Ъ–°) – –і—А—Г–≥–Њ–є –≤–∞–ґ–љ—Л–є –≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ—Л–є —Б–Є–≥–љ–∞–ї–Є–Ј–∞—В–Њ—А —А–µ–∞–Ї—Ж–Є–є, –Ї–Њ—В–Њ—А—Л–є —Г—З–∞—Б—В–≤—Г–µ—В –≤ –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є–Є, –≥–Є–њ–µ—А—В—А–Њ—Д–Є–Є –Є –∞–њ–Њ–њ—В–Њ–Ј–µ. –Ь—Л—И–Є —Б –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ–є —Н–Ї—Б–њ—А–µ—Б—Б–Є–µ–є –†–Ъ–°––±–µ—В–∞ –Є–Ј–Њ—Д–Њ—А–Љ—Л –Ј–∞—Й–Є—Й–µ–љ—Л –Њ—В –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є, –≤ —В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї —Г –Љ—Л—И–µ–є —Б –љ–µ—Е–≤–∞—В–Ї–Њ–є –†–Ъ–°–—Н–њ—Б–Є–ї–Њ–љ –Є–Ј–Њ—Д–Њ—А–Љ—Л —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Б –≤–Њ–Ј—А–∞—Б—В–Њ–Љ –њ–Њ—З–µ—З–љ—Л–є —Д–Є–±—А–Њ–Ј [27,28]. –≠—В–Є —А–µ–Ј—Г–ї—М—В–∞—В—Л –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—В, —З—В–Њ —А–µ–∞–Ї—Ж–Є–Є —Б –†–Ъ–° —П–≤–ї—П—О—В—Б—П –Њ—З–µ–љ—М —Б–ї–Њ–ґ–љ—Л–Љ —А–µ–≥—Г–ї—П—В–Њ—А–љ—Л–Љ –њ—Г—В–µ–Љ –Є –љ—Г–ґ–і–∞—О—В—Б—П –≤ –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–Љ –Є–Ј—Г—З–µ–љ–Є–Є, –њ—А–µ–ґ–і–µ —З–µ–Љ –њ—А–Є–Љ–µ–љ—П—В—М –≤ —В–µ—А–∞–њ–Є–Є –Є—Е –Є–љ–≥–Є–±–Є—В–Њ—А—Л.
–Р–љ—В–∞–≥–Њ–љ–Є—Б—В—Л –∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ–∞
–Э–µ–Њ—Б–њ–Њ—А–Є–Љ–∞ –≤–∞–ґ–љ–∞—П —А–Њ–ї—М –∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ–∞ –Ї–∞–Ї –њ—А–Њ—Д–Є–±—А–Њ–≥–µ–љ–љ–Њ–≥–Њ –∞–≥–µ–љ—В–∞, —Б—В–Є–Љ—Г–ї–Є—А—Г—О—Й–µ–≥–Њ –Љ–Є–Њ–Ї–∞—А–і–Є–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј –Є —Б–µ—А–і–µ—З–љ—Г—О –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М. –Ь–∞–ї–Њ –Є–Ј–≤–µ—Б—В–љ–Њ –Њ –µ–≥–Њ —Г—З–∞—Б—В–Є–Є –≤ —А–∞–Ј–≤–Є—В–Є–Є –њ–∞—В–Њ–ї–Њ–≥–Є–Є –њ–Њ—З–µ–Ї –Є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –±–ї–Њ–Ї–∞—В–Њ—А–Њ–≤ –∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ–∞ –≤ –њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є–Є —Д–Є–±—А–Њ–Ј–∞ –њ–Њ—З–µ–Ї. –Т–≤–µ–і–µ–љ–Є–µ —Б–њ–Є—А–Њ–љ–Њ–ї–∞–Ї—В–Њ–љ–∞ —Г–Љ–µ–љ—М—И–∞–µ—В –њ–Њ—З–µ—З–љ–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ, –∞–Ї—В–Є–≤–љ–Њ—Б—В—М —Д–∞–Ї—В–Њ—А–∞ —А–Њ—Б—В–∞ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є –Є —Б–Є–љ—В–µ–Ј –Ї–Њ–ї–ї–∞–≥–µ–љ–∞, –њ—А–Њ—В–µ–Є–љ—Г—А–Є—О, —Н–Ї—Б–Ї—А–µ—Ж–Є—О –∞–ї—М–±—Г–Љ–Є–љ–∞, —В–∞–Ї–ґ–µ –њ–Њ–і–∞–≤–ї—П–µ—В —А–∞–Ј–≤–Є—В–Є–µ –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –љ–∞ –Ї—А—Л—Б–Є–љ—Л—Е –Љ–Њ–і–µ–ї—П—Е —Б–∞—Е–∞—А–љ–Њ–≥–Њ –і–Є–∞–±–µ—В–∞ 2 —В–Є–њ–∞ [29,30]. –Ъ —В–Њ–Љ—Г –ґ–µ, —Б–њ–Є—А–Њ–љ–Њ–ї–∞–Ї—В–Њ–љ —Г–Љ–µ–љ—М—И–∞–µ—В –і–Є–ї–∞—В–∞—Ж–Є—О –∞—А—В–µ—А–Є–Њ–ї, –∞–њ–Њ–њ—В–Њ–Ј, –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –њ–Њ—З–µ—З–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –Є —В—Г–±—Г–ї–Њ–Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞, —З—В–Њ –њ—А–µ–і—И–µ—Б—В–≤—Г–µ—В —А–∞–Ј–≤–Є—В–Є—О —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є —Ж–Є–Ї–ї–Њ—Б–њ–Њ—А–Є–љ –Р-–Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –љ–µ—Д—А–Њ—В–Њ–Ї—Б–Є—З–љ–Њ—Б—В–Є [31]. –С–ї–Њ–Ї–∞–і–∞ –∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ–Њ–≤—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –љ–µ —Г–ї—Г—З—И–∞–µ—В —Б–Њ—Б—В–Њ—П–љ–Є–µ –њ—А–Є —В—А–∞–≤–Љ–µ –њ–Њ—З–µ–Ї, —Н—В–Њ –±—Л–ї–Њ –≤—Л—П–≤–ї–µ–љ–Њ –љ–∞ –Љ–Њ–і–µ–ї—П—Е —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –≤—Л–Ј–≤–∞–љ–љ–Њ–є –њ—А–Њ—В–µ–Є–љ—Г—А–Є–µ–є —А–µ–љ–∞–ї—М–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–µ–є (–і–Њ–Ї—Б–Њ—А—Г–±–Є—Ж–Є–љ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ—Л–є —Д–Є–±—А–Њ–Ј). –Ґ–∞–Ї–ґ–µ –Є–Р–Я–§ –±—Л–ї–Є —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л –≤ –њ—А–µ–і—Г–њ—А–µ–ґ–і–µ–љ–Є–Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –Є–Ј–Љ–µ–љ–µ–љ–Є–є, –њ—А–µ–і—И–µ—Б—В–≤—Г—О—Й–Є—Е —Д–Є–±—А–Њ–Ј—Г, –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞, —Д–Њ–Ї–∞–ї—М–љ–Њ–≥–Њ –≥–ї–Њ–Љ–µ—А—Г–ї–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –Є –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є. –Ъ–Њ–Љ–±–Є–љ–Є—А–Њ–≤–∞–љ–љ–∞—П —В–µ—А–∞–њ–Є—П –±–ї–Њ–Ї–∞—В–Њ—А–∞–Љ–Є –∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ–∞ –Є –Є–Р–Я–§ –љ–µ –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–µ—В –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л–є –њ—А–Њ—В–µ–Ї—В–Є–≤–љ—Л–є —Н—Д—Д–µ–Ї—В, —З—В–Њ –≥–Њ–≤–Њ—А–Є—В –Њ –і–Њ–≤–Њ–ї—М–љ–Њ –Њ–≥—А–∞–љ–Є—З–µ–љ–љ–Њ–є —А–Њ–ї–Є –∞–ї—М–і–Њ—Б—В–µ—А–Њ–љ–∞ –≤ —А–∞–Ј–≤–Є—В–Є–Є –њ–Њ—З–µ—З–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є [32].
–Р–Ї—В–Є–≤–∞—Ж–Є—П –Ї–Є–љ–Є–љ–Њ–≤—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤
–Я—А–µ–і–њ–Њ–ї–∞–≥–∞–µ—В—Б—П, —З—В–Њ –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–є —Н—Д—Д–µ–Ї—В –Є–Р–Я–§ –Њ–њ–Њ—Б—А–µ–і–Њ–≤–∞–љ –∞–Ї—В–Є–≤–∞—Ж–Є–µ–є –Ї–Є–љ–Є–љ–Њ–≤—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤. –Т –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є–µ —Н—В–Њ–Љ—Г –±–ї–Њ–Ї–∞—В–Њ—А—Л b2–—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ —Б–љ–Є–ґ–∞—О—В —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –Є–Р–Я–§ –њ—А–Є –≥–ї–Њ–Љ–µ—А—Г–ї—П—А–љ–Њ–Љ, –њ–µ—А–Є–≤–∞—Б–Ї—Г–ї—П—А–љ–Њ–Љ –Є —В—Г–±—Г–ї–Њ–Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–Љ —Д–Є–±—А–Њ–Ј–µ, —З—В–Њ –±—Л–ї–Њ –≤—Л—П–≤–ї–µ–љ–Њ –љ–∞ TGRen2 –Љ–Њ–і–µ–ї—П—Е –Ї—А—Л—Б [33]. –Э–∞–Є–±–Њ–ї–µ–µ —П—А–Ї–Њ —Н—В–Њ –њ—А–Њ—Б–ї–µ–ґ–Є–≤–∞–µ—В—Б—П –≤ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е, –≤ –Ї–Њ—В–Њ—А—Л—Е –і–Њ—Б—В–∞–≤–Ї–∞ —З–µ–ї–Њ–≤–µ—З–µ—Б–Ї–Њ–≥–Њ —В–Ї–∞–љ–µ–≤–Њ–≥–Њ –Ї–∞–ї–ї–Є–Ї—А–µ–Є–љ–Њ–≤–Њ–≥–Њ –≥–µ–љ–∞ —Г–ї—Г—З—И–Є–ї–∞ –њ–Њ—З–µ—З–љ—Г—О —Д—Г–љ–Ї—Ж–Є—О –Є —Б–љ–Є–Ј–Є–ї–∞ –Љ–Њ–љ–Њ—Ж–Є—В–∞—А–љ–Њ––Љ–∞–Ї—А–Њ—Д–∞–≥–∞–ї—М–љ—Г—О –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є—О, —Н–Ї—Б–њ—А–µ—Б—Б–Є—О —Е–µ–Љ–Њ–∞—В—В—А–∞–Ї—В–∞–љ—В–љ—Л—Е –њ–µ–њ—В–Є–і–1 –Љ–Њ–љ–Њ—Ж–Є—В–Њ–≤ –Є –Ї–ї–µ—В–Њ—З–љ—Л–є –∞–њ–Њ–њ—В–Њ–Ј –њ—А–Є –≥–µ–љ—В–∞–Љ–Є—Ж–Є–љ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –љ–µ—Д—А–Њ—В–Њ–Ї—Б–Є—З–љ–Њ—Б—В–Є [34]. –Р–љ–∞–ї–Њ–≥–Є—З–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ, –і–Њ—Б—В–∞–≤–Ї–∞ —З–µ–ї–Њ–≤–µ—З–µ—Б–Ї–Њ–≥–Њ —В–Ї–∞–љ–µ–≤–Њ–≥–Њ –Ї–∞–ї–ї–Є–Ї—А–µ–Є–љ–Њ–≤–Њ–≥–Њ –≥–µ–љ–∞ –Є–ї–Є –Ї–Є–љ–Є–љ–Њ–≤–∞—П –Є–љ—Д—Г–Ј–Є—П –Њ—Б–ї–∞–±–ї—П—О—В –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ –њ–Њ—З–µ–Ї –љ–∞ Dahl––Љ–Њ–і–µ–ї—П—Е –Ї—А—Л—Б. –Ю–±–∞ –њ–Њ–і—Е–Њ–і–∞ —Г–ї—Г—З—И–∞—О—В –њ–Њ—З–µ—З–љ—Л–µ —Б—В—А—Г–Ї—В—Г—А—Г –Є —Д—Г–љ–Ї—Ж–Є–Є, —З—В–Њ –њ—А–Њ—П–≤–ї—П–µ—В—Б—П –≤ —Г–Љ–µ–љ—М—И–µ–љ–Є–Є –Њ—В–ї–Њ–ґ–µ–љ–Є—П –Ї–Њ–ї–ї–∞–≥–µ–љ–∞, –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –Љ–Њ–љ–Њ—Ж–Є—В–∞—А–љ–Њ––Љ–∞–Ї—А–Њ—Д–∞–≥–∞–ї—М–љ–Њ–≥–Њ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—П, –Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –∞–њ–Њ–њ—В–Њ–Ј–∞ –≤ —В—А—Г–±–Њ—З–Ї–∞—Е, —Б–љ–Є–ґ–µ–љ–Є–Є –ї—О–Љ–Є–љ–∞–ї—М–љ–Њ–є –њ—А–Њ—В–µ–Є–љ–Њ–≤–Њ–є —Д–Њ—А–Љ–∞—Ж–Є–Є, –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є —Б —П–≤–љ—Л–Љ –≤–ї–Є—П–љ–Є–µ–Љ –љ–∞ –Р–Ф. –Я—А–µ–і–њ–Њ–ї–∞–≥–∞–µ–Љ—Л–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –Ј–∞–Ї–ї—О—З–∞–µ—В—Б—П –≤ —В–Њ–Љ, —З—В–Њ –Є–љ—Д—Г–Ј–Є—П –Ї–Є–љ–Є–љ–Њ–≤ –Љ–Њ–ґ–µ—В –љ–∞–њ—А—П–Љ—Г—О –Ј–∞—Й–Є—Й–∞—В—М –Њ—В –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ —Б–Њ–ї—М—О –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –њ–Њ—З–µ–Ї –±–µ–Ј —Б–љ–Є–ґ–µ–љ–Є—П –Р–Ф, —Б –њ–Њ–Љ–Њ—Й—М—О –Є–љ–≥–Є–±–Є—А–Њ–≤–∞–љ–Є—П –∞–њ–Њ–њ—В–Њ–Ј–∞, –≤–Њ—Б–њ–∞–ї–µ–љ–Є—П, —Д–Є–±—А–Њ–Ј–∞ —З–µ—А–µ–Ј –њ–Њ–і–∞–≤–ї–µ–љ–Є–µ –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ —Б—В—А–µ—Б—Б–∞, TGFb- –Є –Ь–Р–†–Ъ-–∞–Ї—В–Є–≤–∞—Ж–Є–Є [7,35,36].
–°—В–∞—В–Є–љ—Л
–Я–Њ—Б–ї–µ–і–љ–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—В, —З—В–Њ —Б—В–∞—В–Є–љ—Л –њ—А–µ–њ—П—В—Б—В–≤—Г—О—В —А–∞–Ј–≤–Є—В–Є—О —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ—З–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –≤–љ–µ –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В –Є—Е –≥–Є–њ–Њ–ї–Є–њ–Є–і–µ–Љ–Є—З–µ—Б–Ї–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П. –Я–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ —А–∞–Ј–ї–Є—З–љ—Л–µ —Б—В–∞—В–Є–љ—Л –Є–љ–≥–Є–±–Є—А—Г—О—В –њ–µ—А–µ—Е–Њ–і –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е —В—Г–±—Г–ї—П—А–љ—Л—Е —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –≤ –Љ–µ–Ј–µ–љ—Е–Є–Љ–∞–ї—М–љ—Л–µ, —Б–Њ—Е—А–∞–љ—П—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—О —В–∞–Ї–Є—Е –Љ–∞—А–Ї–µ—А–Њ–≤ —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї, –Ї–∞–Ї –Х––Ї–∞–і–µ—А–Є–љ –Є —Ж–Є—В–Њ–Ї–µ—А–∞—В–Є–љ–19, –Є —Б–љ–Є–ґ–∞—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—О FnEDA. –Ф–µ–є—Б—В–≤–Є–µ –Љ–∞—А–Ї–µ—А–∞ –Љ–Є–Њ—Д–Є–±—А–Њ–±–ї–∞—Б—В–љ–Њ–≥–Њ —Д–µ–љ–Њ—В–Є–њ–∞ [37]. –Ы–µ—З–µ–љ–Є–µ —Б—В–∞—В–Є–љ–∞–Љ–Є —В–∞–Ї–ґ–µ –±—Л–ї–Њ –Є–Ј—Г—З–µ–љ–Њ –љ–∞ –Љ–Њ–і–µ–ї–Є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–≤–љ–Њ–є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є (–њ—А–Є —Б–Њ–ї–µ–≤–Њ–є –љ–∞–≥—А—Г–Ј–Ї–µ). –Я—А–Є —Н—В–Њ–Љ —Г –ґ–Є–≤–Њ—В–љ—Л—Е —А–∞–Ј–≤–Є–ї–Є—Б—М –∞—А—В–µ—А–Є–∞–ї—М–љ–∞—П –≥–Є–њ–µ—А—В–µ–љ–Ј–Є—П, –њ—А–Њ—В–µ–Є–љ—Г—А–Є—П, –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є—П –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є, –њ—А–Њ–Є–Ј–Њ—И–ї–Њ —Б–љ–Є–ґ–µ–љ–Є–µ —З–Є—Б–ї–∞ –њ–Њ–і–Њ—Ж–Є—В–Њ–≤ –Є –њ–Њ—П–≤–Є–ї—Б—П –≤—Л—А–∞–ґ–µ–љ–љ—Л–є —Д–Є–±—А–Њ–Ј. –≠—В–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–ї–Є—Б—М –і–Є—Б–±–∞–ї–∞–љ—Б–Њ–Љ –≤ –њ–ї–∞–Ј–Љ–Є–љ–Њ–≥–µ–љ––њ–ї–∞–Ј–Љ–Є–љ–Њ–≤–Њ–є –Є –Љ–µ—В–∞–ї–ї–Њ–њ—А–Њ—В–µ–∞–Ј–љ–Њ–є —Б–Є—Б—В–µ–Љ–∞—Е. –Ф–ї–Є—В–µ–ї—М–љ–∞—П —В–µ—А–∞–њ–Є—П –≥–Є–і—А–Њ—Д–Є–ї—М–љ—Л–Љ —А–Њ–Ј—Г–≤–∞—Б—В–∞—В–Є–љ–Њ–Љ –Њ–Ї–∞–Ј—Л–≤–∞–ї–∞ –љ–µ—Д—А–Њ–њ—А–Њ—В–µ–Ї—В–Є–≤–љ—Л–є —Н—Д—Д–µ–Ї—В, –њ—А–µ–і—Г–њ—А–µ–ґ–і–∞–ї–∞ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ–ї–∞–Ј–Љ–Є–љ–∞ –Є –Љ–µ—В–∞–ї–ї–Њ–њ—А–Њ—В–µ–Є–љ–∞–Ј–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞ (–Ь–Ь–†), –љ–µ–Ј–∞–≤–Є—Б–Є–Љ–Њ –Њ—В –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П [38].
–§–∞–Ї—В–Њ—А—Л –і–µ–≥—А–∞–і–∞—Ж–Є–Є
—Н–Ї—Б—В—А–∞—Ж–µ–ї–ї—О–ї—П—А–љ–Њ–≥–Њ –Љ–∞—В—А–Є–Ї—Б–∞
–Э–∞ —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –Љ–Њ–і–µ–ї—П—Е –і–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –∞–Ї—В–Є–≤–∞—Ж–Є—П –Ь–Ь–† –Є–≥—А–∞–µ—В –≤–∞–ґ–љ—Г—О —А–Њ–ї—М, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –±–ї–Њ–Ї–Є—А—Г–µ—В—Б—П —А–∞–Ј–≤–Є—В–Є–µ –њ–Њ—З–µ—З–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ –њ—А–Є –і–µ—Д–Є—Ж–Є—В–µ –Њ–Ї—Б–Є–і–∞ –∞–Ј–Њ—В–∞ [1]. –° –љ–∞—И–µ–є —В–Њ—З–Ї–Є –Ј—А–µ–љ–Є—П, —А–µ–≥—Г–ї–Є—А—Г–µ–Љ–∞—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—П –Ь–Ь–† —Б–≤—П–Ј–∞–љ–∞ —Б –Ї–Њ–Љ–њ–µ–љ—Б–∞—В–Њ—А–љ—Л–Љ–Є –Љ–µ—Е–∞–љ–Є–Ј–Љ–∞–Љ–Є, –Њ–≥—А–∞–љ–Є—З–Є–≤–∞—О—Й–Є–Љ–Є –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –Љ–∞—В—А–Є–Ї—Б–∞. –Ф—А—Г–≥–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—В, –Њ–і–љ–∞–Ї–Њ, —З—В–Њ –њ–Њ–≤—Л—И–µ–љ–љ–∞—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—П –∞–Ї—В–Є–≤–љ–Њ–≥–Њ –Ь–Ь–†2 –≤ –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Ї–∞–љ–∞–ї—М—Ж–∞—Е –Њ—В—А–∞–ґ–∞–µ—В —В—Г–±—Г–ї—П—А–љ—Г—О –∞—В—А–Њ—Д–Є—О, —Д–Є–±—А–Њ–Ј –Є –њ–Њ—З–µ—З–љ—Л–µ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П [39].
–Ґ–∞–Ї–Њ–є —Б–њ–Њ—А –Љ–Њ–≥—Г—В —А–∞–Ј—А–µ—И–Є—В—М —А–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –љ–∞ –ґ–Є–≤–Њ—В–љ—Л—Е –Љ–Њ–і–µ–ї—П—Е, –Є–Љ–Є—В–Є—А—Г—О—Й–Є—Е –≥–µ–љ–µ—В–Є—З–µ—Б–Ї—Г—О —Д–Њ—А–Љ—Г —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ—З–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є – —Б–Є–љ–і—А–Њ–Љ Alport [40]. –§–∞—А–Љ–∞–Ї–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є–љ–≥–Є–±–Є—А–Њ–≤–∞–љ–Є–µ —Д–µ—А–Љ–µ–љ—В–љ–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ь–Ь–† –і–Њ –і–µ–±—О—В–∞ –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є –Є —Б—В—А—Г–Ї—В—Г—А–љ—Л—Е –і–µ—Д–µ–Ї—В–Њ–≤ –Ї–ї—Г–±–Њ—З–Ї–Њ–≤ –њ—А–Є–≤–µ–ї–Њ –Ї –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–Љ—Г —Г–ї—Г—З—И–µ–љ–Є—О —В–µ—З–µ–љ–Є—П –±–Њ–ї–µ–Ј–љ–Є, —З—В–Њ –±—Л–ї–Њ —Б–≤—П–Ј–∞–љ–Њ —Б –Њ—В—Б—А–Њ—З–Ї–Њ–є —А–∞–Ј–≤–Є—В–Є—П –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є, –љ–Њ –≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ – —Б –њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ –њ—А–Њ–і–Њ–ї–ґ–Є—В–µ–ї—М–љ–Њ—Б—В–Є –ґ–Є–Ј–љ–Є. –Т –њ—А–Њ—В–Є–≤–Њ–њ–Њ–ї–Њ–ґ–љ–Њ—Б—В—М —Н—В–Њ–Љ—Г –Є–љ–≥–Є–±–Є—А–Њ–≤–∞–љ–Є–µ –Ь–Ь–† –њ–Њ—Б–ї–µ –љ–∞—З–∞–ї–∞ –њ—А–Њ—В–µ–Є–љ—Г—А–Є–Є –њ—А–Є–≤–µ–ї–Њ –Ї —Г—Б–Ї–Њ—А–µ–љ–Є—О —В–µ—З–µ–љ–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, —З—В–Њ, –≤ —Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –±—Л–ї–Њ –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–Њ —Б –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ–Љ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ –Є —А–∞–љ–љ–µ–є —Б–Љ–µ—А—В–љ–Њ—Б—В—М—О.
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –∞–Ї—В–Є–≤–∞—Ж–Є—П –Ь–Ь–† –Љ–Њ–ґ–µ—В –Њ–Ї–∞–Ј—Л–≤–∞—В—М –і–≤–Њ–є–љ–Њ–є —Н—Д—Д–µ–Ї—В –љ–∞ —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ—З–µ–Ї, —З—В–Њ –Ј–∞–≤–Є—Б–Є—В –Њ—В —Б—В–∞–і–Є–Є –њ–Њ—З–µ—З–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Ю—З–µ–≤–Є–і–љ–Њ –њ—А–Њ–±–ї–µ–Љ–∞ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П —Д–∞–Ї—В–Њ—А–Њ–≤, –љ–∞—Ж–µ–ї–µ–љ–љ—Л—Е –љ–∞ –Љ–∞—В—А–Є–Ї—Б––і–µ–≥—А–∞–і–Є—А—Г—О—Й–Є–µ —Д–µ—А–Љ–µ–љ—В—Л, –≤ –Ї–∞—З–µ—Б—В–≤–µ —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б—А–µ–і—Б—В–≤–∞, —З—В–Њ, –≤ —Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –њ–Њ–Љ–Њ–ґ–µ—В –ї—Г—З—И–µ –њ–Њ–љ—П—В—М –Є—Е –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –≤ —А–∞–Ј–ї–Є—З–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А–∞—Е –њ–Њ—З–Ї–Є.
–Ш–љ–≥–Є–±–Є—А–Њ–≤–∞–љ–Є–µ —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –Ї–Њ–ї–ї–∞–≥–µ–љ–∞
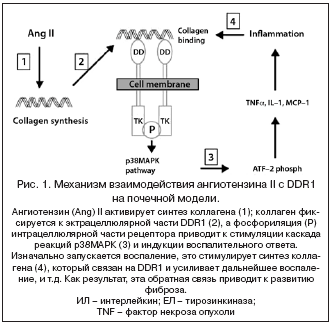
–°—А–µ–і–Є —Б–Є—Б—В–µ–Љ, –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤—Г—О—Й–Є—Е —Б —Н–Ї—Б—В—А–∞—Ж–µ–ї–ї—О–ї—П—А–љ—Л–Љ –Љ–∞—В—А–Є–Ї—Б–Њ–Љ –Є –і–µ–є—Б—В–≤—Г—О—Й–Є—Е –Ї–∞–Ї —А–µ—Ж–µ–њ—В–Њ—А—Л –Ї–Њ–ї–ї–∞–≥–µ–љ–∞, –≤—Л–і–µ–ї—П—О—В DDR1 (discoidin domain receptors 1). –≠—В–Є —А–µ—Ж–µ–њ—В–Њ—А—Л –Њ—Б–Њ–±–µ–љ–љ—Л —В–µ–Љ, —З—В–Њ –њ—А–Є —Б–≤—П–Ј—Л–≤–∞–љ–Є–Є —Б –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–Љ –Њ–љ–Є –љ–∞—З–Є–љ–∞—О—В –і–µ–є—Б—В–≤–Њ–≤–∞—В—М, –Ї–∞–Ї —А–µ—Ж–µ–њ—В–Њ—А—Л —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Њ—Б—В–∞: –Њ–љ–Є –і–Є–Љ–µ—А–Є–Ј—Г—О—В—Б—П, —Д–Њ—Б—Д–Њ—А–Є–ї–Є—А—Г—О—В—Б—П —В–Є—А–Њ–Ј–Є–љ–Ї–Є–љ–∞–Ј–Њ–є –Є —Н—В–∞ —Д–Њ—Б—Д–Њ—А–Є–ї—П—Ж–Є—П —Б—В–Є–Љ—Г–ї–Є—А—Г–µ—В —А–µ–∞–Ї—Ж–Є–Є —А38–Ь–Р–†–Ъ [41]. –Ф–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П in vitro –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ DDR1 –љ–µ–Њ–±—Е–Њ–і–Є–Љ –і–ї—П —Б–Њ–Ј—А–µ–≤–∞–љ–Є—П –Є –і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–Ї–Є –Љ–Њ–љ–Њ—Ж–Є—В–Њ–≤ –Є –Љ–∞–Ї—А–Њ—Д–∞–≥–Њ–≤ –Є –Љ–Њ–ґ–µ—В –±—Л—В—М –њ—А–Њ–Љ–µ–ґ—Г—В–Њ—З–љ—Л–Љ –Ј–≤–µ–љ–Њ–Љ –≤ –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–Љ –Њ—В–≤–µ—В–µ [42]. –Ь—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–ї–Є —А–∞–Ј–≤–Є—В–Є–µ –Є –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–є —Б–Њ—Б—Г–і–Њ–≤ –Є –Ї–ї—Г–±–Њ—З–Ї–Њ–≤ —Г DDR1–—З–Є—Б—В—Л—Е –Љ—Л—И–µ–є –љ–∞ —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ–Њ–є –Љ–Њ–і–µ–ї–Є –љ–µ—Д—А–Њ–њ–∞—В–Є–Є, –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –≤–≤–µ–і–µ–љ–Є–µ–Љ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–∞ II. –£ –Љ—Л—И–µ–є –і–Є–Ї–Њ–≥–Њ —В–Є–њ–∞ —А–∞–Ј–≤–Є–ї–∞—Б—М –≥–Є–њ–µ—А—В–µ–љ–Ј–Є—П, –њ–µ—А–Є–≤–∞—Б–Ї—Г–ї—П—А–љ–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ, –≥–ї–Њ–Љ–µ—А—Г–ї—П—А–љ—Л–є —Б–Ї–ї–µ—А–Њ–Ј –Є –њ—А–Њ—В–µ–Є–љ—Г—А–Є—П. –Э–µ—Б–Љ–Њ—В—А—П –љ–∞ —Б—Е–Њ–і–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ –Р–Ф —Г –Љ—Л—И–µ–є —Б –і–µ—Д–Є—Ж–Є—В–Њ–Љ DDR1, –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –њ—А–Є –≥–ї–Њ–Љ–µ—А—Г–ї—П—А–љ–Њ–Љ —Д–Є–±—А–Њ–Ј–µ –Є –≤–Њ—Б–њ–∞–ї–µ–љ–Є–Є –±—Л–ї–Є –Ї–≤–∞–Ј–Є––Њ—В—Б—Г—В—Б—В–≤—Г—О—Й–Є–Љ–Є, –∞ –њ—А–Њ—В–µ–Є–љ—Г—А–Є—П –±—Л–ї–∞ –≤ –Њ—Б–љ–Њ–≤–љ–Њ–Љ –њ—А–µ–і—Г–њ—А–µ–ґ–і–µ–љ–∞. –Ю–Ї—А–∞—И–Є–≤–∞–љ–Є–µ –ї–Є–Љ—Д–Њ—Ж–Є—В–Њ–≤, –Љ–∞–Ї—А–Њ—Д–∞–≥–Њ–≤ –Є –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ I –Є IV —В–Є–њ–Њ–≤ –±—Л–ї–Њ —П–≤–љ–Њ –Ј–∞–Љ–µ—В–љ–Њ –≤ –њ–Њ—З–µ—З–љ–Њ–Љ –Ї–Њ—А—В–µ–Ї—Б–µ —Г –ґ–Є–≤–Њ—В–љ—Л—Е –і–Є–Ї–Њ–≥–Њ —В–Є–њ–∞ –Є –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є –љ–µ—Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —Г DDR1–—З–Є—Б—В—Л—Е –Љ—Л—И–µ–є [43].
–Ь—Л –њ—А–µ–і–њ–Њ–ї–∞–≥–∞–µ–Љ, —З—В–Њ –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ II –∞–Ї—В–Є–≤–Є—А—Г–µ—В—Б—П –њ—А–Є —Б–Є–љ—В–µ–Ј–µ –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ (—А–Є—Б. 1). –Ъ–Њ–ї–ї–∞–≥–µ–љ —Б–≤—П–Ј—Л–≤–∞–µ—В—Б—П –Њ–і–Є–љ —А–∞–Ј –≤ —Н–Ї—Б—В—А–∞—Ж–µ–ї–ї—О–ї—П—А–љ–Њ–Љ —З–∞—Б—В–Є DDR1, –Є–љ—В—А–∞—Ж–µ–ї–ї—О–ї—П—А–љ–∞—П —З–∞—Б—В—М —А–µ—Ж–µ–њ—В–Њ—А–∞ –њ—А–Є–≤–Њ–і–Є—В –Ї –Є–љ–і—Г–Ї—Ж–Є–Є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ –Њ—В–≤–µ—В–∞. –Т–Њ—Б–њ–∞–ї–µ–љ–Є–µ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ —Б–Є–љ—В–µ–Ј–∞ –Ї–Њ–ї–ї–∞–≥–µ–љ–∞ –Є —Б–≤—П–Ј—Л–≤–∞–љ–Є–µ–Љ, —З—В–Њ, –≤ —Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, —Г—Б–Є–ї–Є–≤–∞–µ—В –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ –Є —В–µ—З–µ–љ–Є–µ –Є–і–µ—В –њ–Њ –Ј–∞–Љ–Ї–љ—Г—В–Њ–Љ—Г –Ї—А—Г–≥—Г, –њ—А–Є–≤–Њ–і—П –Ї —А–∞–Ј–≤–Є—В–Є—О —Д–Є–±—А–Њ–Ј–∞. –Р–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ II –Є–љ–Є—Ж–Є–Є—А—Г–µ—В –∞–Ї—В–Є–≤–∞—Ж–Є—О DDR1, –љ–Њ —Б–љ–∞—З–∞–ї–∞ –∞–Ї—В–Є–≤–∞—Ж–Є—П DDR1 –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –ї–Њ–Ї–∞–ї—М–љ–Њ. –Т–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є–µ –Љ–µ–ґ–і—Г DDR1, –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ–Љ, –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–Љ –Є —Д–Є–±—А–Њ–Ј–Њ–Љ —П–≤–ї—П–µ—В—Б—П —Б–∞–Љ–Њ–њ–Њ–і–і–µ—А–ґ–Є–≤–∞—О—Й–Є–Љ—Б—П –њ—А–Њ—Ж–µ—Б—Б–Њ–Љ –Є –љ–µ –љ—Г–ґ–і–∞–µ—В—Б—П –≤ –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–Љ –і–µ–є—Б—В–≤–Є–Є –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–∞ II.
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ: –≤–Ј–≥–ї—П–і –Є–Ј –±—Г–і—Г—Й–µ–≥–Њ
–Т —В–∞–±–ї–Є—Ж–µ 1 –Њ–±–Њ–±—Й–µ–љ—Л —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –≤ –Ї–Њ—В–Њ—А—Л—Е –≤—Л—П–≤–ї–µ–љ —А–µ–≥—А–µ—Б—Б –њ–Њ—З–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є. –°–Њ–≤–µ—А—И–µ–љ–љ–Њ —П—Б–љ–Њ, —З—В–Њ –≤ –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ –Є—Б–њ—Л—В–∞–љ–Є–є –љ–∞–±–ї—О–і–∞–µ–Љ—Л–є —А–µ–≥—А–µ—Б—Б –±—Л–ї –≤—Л–Ј–≤–∞–љ –±–ї–Њ–Ї–∞–і–Њ–є —А–µ–љ–Є–љ––∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–Њ–≤–Њ–є —Б–Є—Б—В–µ–Љ—Л. –Т –њ–Њ—Б–ї–µ–і—Г—О—Й–Є–µ –≥–Њ–і–∞ —Б–њ–Є—Б–Њ–Ї –Љ–µ–і–Є–∞—В–Њ—А–Њ–≤ –њ—А–Њ—Д–Є–±—А–Њ–≥–µ–љ–љ–Њ–≥–Њ –і–µ–є—Б—В–≤–Є—П –∞–љ–≥–Є–Њ—В–µ–љ–Ј–Є–љ–∞ II (—А–Є—Б. 2) –±—Г–і–µ—В —А–∞—Б—В–Є, –±—Г–і—Г—В –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ—Л –љ–Њ–≤—Л–µ –Љ–Є—И–µ–љ–Є. –С—Г–і—Г—Й–µ–µ –Ј–∞ –њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є, —Б–њ–Њ—Б–Њ–±–љ—Л–Љ–Є –Є–љ–≥–Є–±–Є—А–Њ–≤–∞—В—М —В–∞–Ї–Є–µ –Љ–∞—В–µ—А–Є–∞–ї—Л, –Ї–∞–Ї DDR1, –Ї–Њ—В–Њ—А—Л–є —П–≤–ї—П–µ—В—Б—П –Љ–µ–і–Є–∞—В–Њ—А–Њ–Љ –Њ–і–љ–Њ–≤—А–µ–Љ–µ–љ–љ–Њ –њ—А–Є –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є–Є, —Д–Є–±—А–Њ–Ј–µ –Є –≤–Њ—Б–њ–∞–ї–µ–љ–Є–Є. –Э–µ–Њ—Б–њ–Њ—А–Є–Љ–∞ –≤–∞–ґ–љ–Њ—Б—В—М –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–Њ —А–∞–љ–љ–µ–≥–Њ –љ–∞—З–∞–ї–∞ –ї–µ—З–µ–љ–Є—П, –і–Њ –љ–µ–Њ–±—А–∞—В–Є–Љ–Њ–≥–Њ —Г—Е—Г–і—И–µ–љ–Є—П –њ–Њ—З–µ—З–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Ї—А–∞–є–љ–µ –≤–∞–ґ–љ–Њ –Є–Ј—Г—З–∞—В—М –ї–µ–≥–Ї–Њ –≤—Л–і–µ–ї—П–µ–Љ—Л–µ –Љ–∞—А–Ї–µ—А—Л (–≤ –Љ–Њ—З–µ –Є–ї–Є –њ–ї–∞–Ј–Љ–µ), –њ—А–Њ–≥–љ–Њ–Ј–Є—А—Г—О—Й–Є–µ —А–∞–Ј–≤–Є—В–Є–µ –њ–Њ—З–µ—З–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є. –Ъ–Њ–Љ–±–Є–љ–Є—А–Њ–≤–∞–љ–Є–µ –і–∞–љ–љ—Л—Е –њ–Њ–і—Е–Њ–і–Њ–≤ –њ—А–Њ–Є–Ј–≤–µ–і–µ—В —А–µ–≤–Њ–ї—О—Ж–Є—О –≤ –ї–µ—З–µ–љ–Є–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ—З–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є, –њ–µ—А–µ–≤–µ–і—П —Н—В—Г –њ–∞—В–Њ–ї–Њ–≥–Є—О –≤ —А–∞–Ј—А—П–і –Њ–±—А–∞—В–Є–Љ—Л—Е –Є –њ–Њ–ї–љ–Њ—Б—В—М—О –Є–Ј–ї–µ—З–Є–Љ—Л—Е.
–†–µ—Д–µ—А–∞—В –њ–Њ–і–≥–Њ—В–Њ–≤–ї–µ–љ –≠.–†. –Т–µ–ї–Є–Ї–Њ–≤–Њ–є
–њ–Њ –Љ–∞—В–µ—А–Є–∞–ї–∞–Љ —Б—В–∞—В—М–Є Christos Chatziantoniou –Є Jean–Claude Dussaule «Is kidney injury a reversible process?», Current Opinion in Nephrology and Hypertension, 2008, 17: 76–81


–Ы–Є—В–µ—А–∞—В—Г—А–∞
1. Boffa JJ, Ying L, Placier S, et al. Regression of renal vascular and glomerular fibrosis: Role of angiotensin II receptor antagonism and metalloproteinases. J Am Soc Nephrol 2003; 14:1132–1144.
2. Adamczak M, Gross ML, Krtil J, et al. Reversal of glomerulosclerosis after highdose enalapril treatment in subtotally nephrectomized rats. J Am Soc Nephrol 2003; 14:2833–2842.
3. Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860.
4. Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
5. Parving HH, Lehnert H, Brochner-Mortensen J, et al. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001; 345:870–878.
6. Placier S, Boffa JJ, Dussaule JC, Chatziantoniou C. Reversal of renal lesions following interruption of nitric oxide synthesis inhibition in transgenic mice. Nephrol Dial Transplant 2006; 21:881–888.
7. Bledsoe G, Shen B, Yao Y, et al. Reversal of renal fibrosis, inflammation, and glomerular hypertrophy by kallikrein gene delivery. Hum Gene Ther 2006; 17:545–555.
8. Chow FY, Nikolic-Patzrson DJ, Ozols E, et al. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocintreated mice. Kidney Int 2006; 69:73–80.
9. Yamahana J, Wada T, Furuichi K, et al. TAK-603, an anti-inflammatory compound, reduces crescentic glomerulonephritis and preserves renal function in WKY rats. Nephrol Dial Transplant 2006; 21:2736–2744.
10. Wu MJ, Wen MC, Chiu YT, et al. Rapamycin attenuates unilateral ureteral obstruction-induced renal fibrosis. Kidney Int 2006; 69:2029–2036.
11. Kuwabara N, Tamada S, Iwai T, et al. Attenuation of renal fibrosis by curcumin in rat obstructive nephropathy. Urology 2006; 67:440–446.
12. Wang S, de Caestecker M, Kopp J, et al. Renal bone morphogenetic protein-7 protects against diabetic nephropathy. J Am Soc Nephrol 2006; 17:2504–2512.
13. Sugimoto H, Grahovac G, Zeisberg M, Kalhuri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes 2007; 56:1825–1833.
14. Liu Y, Yang J. Hepatocyte growth factor: new arsenal in the fights against renal fibrosis? Kidney Int 2006; 70:238–240.
15. Herrero-Fresneda I, Torras J, Franquesa M, et al. HGF gene therapy attenuates renal allograft scarring by preventing the profibrotic inflammatory-induced mechanisms. Kidney Int 2006; 70:265–274.
16. Moon JA, Kim HT, Cho IS, et al. IN-1130, a novel transforming growth factorbeta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int 2006; 70:1234–1243.
17. Kelly DJ, Zhang Y, Cox AJ, Gilbert RE. Combination therapy with tranilast and angiotensin-converting enzyme inhibition provides additional renoprotection in the remnant kidney model. Kidney Int 2006; 69:1954–1960.
18. Soma J, Sato K, Saito H, Tsuchiya Y. Effect of tranilast in early-stage diabetic nephropathy. Nephrol Dial Transplant 2006; 21:2795–2799.
19. El Chaar M, Chen J, Seshan SV, et al. Effect of combination therapy with enalapril and the TGF-beta antagonist 1D11 in unilateral ureteral obstruction. Am J Physiol Renal Physiol 2007; 292:F1291–1301.
20. Hwang M, Kim HJ, Noh HJ, et al. TGF-beta1 siRNA suppresses the tubulointerstitial fibrosis in the kidney of ureteral obstruction. Exp Mol Pathol 2006; 81:48–54.
21. Franc.ois H, Placier S, Flamant M, et al. Prevention of renal vascular and glomerular fibrosis by epidermal growth factor receptor inhibition. FASEB J 2004; 18:926–928.
22. Schellings MW, Baumann M, van Leeuwen RE, et al. Imatinib attenuates end-organ damage in hypertensive homozygous TGR(mRen2)27 rats. Hypertension 2006; 47:467–474.
23. Ostendorf T, Rong S, Boor P, et al. Antagonism of PDGF-D by human antibody CR002 prevents renal scarring in experimental glomerulonephritis. J Am Soc Nephrol 2006; 17:1054–1062.
24. Boor P, Konieczny A, Villa L, et al. PDGF-D inhibition by CR002 ameliorates tubulointerstitial fibrosis following experimental glomerulonephritis. Nephrol Dial Transplant 2007; 22:1323–1331.
25. Wada T, Azuma H, Furiuchi K, et al. Reduction in chronic allograft nephropathy by inhibition of p38 mitogen-activated protein kinase. Am J Nephrol 2006; 26:319–325.
26. Park JK, Fischer R, Dechend R, et al. p38 mitogen-activated protein kinase inhibition ameliorates angiotensin II-induced target organ damage. Hypertension 2007; 49:481–489.
27. Meier M, Park JK,Overhau D, et al. Deletion of protein kinase C-beta isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocininduced diabetic mouse model. Diabetes 2007; 56:346–354.
28. Meier M, Menne J, Park JK, et al. Deletion of protein kinase C-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol 2007; 18:1190–1198.
29. Han KH, Kang YS, Han SY, et al. Spironolactone ameliorates renal injury and connective tissue growth factor expression in type II diabetic rats. Kidney Int 2006; 70:111–120.
30. Han SY, Kim CH, Kim HS, et al. Spironolactone prevents diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. J Am Soc Nephrol 2006; 17:1362–1372.
31. Perez-Rojas J, Blanco JA, Cruz C, et al. Mineralocorticoid receptor blockade confers renoprotection in preexisting chronic cyclosporine nephrotoxicity. Am J Physiol Renal Physiol 2007; 292:F131–139.
32. Kramer AB, van der Meulen EF, Hamming I, et al. Effect of combining ACE inhibition with aldosterone blockade on proteinuria and renal damage in experimental nephrosis. Kidney Int 2007; 71:417–424.
33. Sceccia TM, Belloni AS, Guidolin D, et al. The renal antifibrotic effect of angiotensin-converting enzyme inhibition involve bradykinin B2 receptor activation in angiotensin II-dependent hypertension. J Hypertens 2006; 24:1419–1427.
34. Bledsoe G, Crickman S, Mao J, et al. Kallikrein/kinin protects against gentamicin-induced nephrotoxicity by inhibition of inflammation and apoptosis. Nephrol Dial Transplant 2006; 21:624–633.
35. Chao J, Li HJ, Yao YY, et al. Kinin infusion prevents renal inflammation, apoptosis, and fibrosis via inhibition of oxidative stress and mitogen-activated protein kinase activity. Hypertension 2007; 49:490–497.
36. Chao J, Bledsoe G, Yin H, Chao L. The tissue kallikrein-kinin system protects against cardiovascular and renal diseases and ischemic stroke independently of blood pressure reduction. Biol Chem 2006; 387:665–675.
37. Patel S, Mason R, Suzuki J, et al. Inhibitory effect of statins on renal epithelial to mesenchymal transition. Am J Nephrol 2006; 26:381–387.
38. Gianella A, Nobili E, Abbate M, et al. Rosuvastatin treatment prevents progressive kidney inflammation and fibrosis in stroke-prone rats. Am J Pathol 2007; 170:1165–1177.
39. Cheng S, Pollock AS, Mahimkar R, et al. Matrix metalloproteinase 2 and basement membrane integrity: a unifying mechanism for progressive renal injury. FASEB J 2006; 20:1898–1900.
40. Zeisberg M, Khurana M, Rao VH, et al. Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med 2006; 3:e100.
41. Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell 1997; 1:13–23.
42. Matsuyama W, Kamohara H, Galligan C, et al. Interaction of discoidin domain receptor 1 isoform b (DDR1b) with collagen activates p38 mitogen-activated protein kinase and promotes differentiation of macrophages. FASEB J 2003; 17:1286–1288.
43. Flamant M, Placier S, Rodenas A, et al. Discoidin domain receptor 1 null mice are protected against hypertension-induced renal disease. J Am Soc Nephrol 2006; 17:3374–3381.



