




Высокая встречаемость эпилепсий в общей популяции, составляющая по данным большинства эпидемиологических исследований 5–8 на 1000 [32,68], течение заболевания, нередко приводящее к психосоциальной дезадаптации и инвалидизации больного, определяют особую актуальность различных аспектов фармакотерапии – основного вида лечения эпилепсий взрослых.
Реактивные и неспровоцированные припадки
Перед началом противоэпилептического лечения необходимо решить вопрос о том, являются ли припадки реактивными (симптоматическими), связанными с воздействием токсических, инфекционных или метаболических факторов, или неспровоцированными. В первом случае антиэпилептические препараты (АЭП) назначаются лишь для купирования припадка в остром периоде [64]. У больных с однократными неспровоцированными эпилептическими припадками (ЭП), не имеющих факторов риска развития эпилепсии, очаговой неврологической симптоматики и патологических изменений на ЭЭГ, дальнейшее течение заболевания различно. Только у 1/4 из них позже развивается эпилепсия, при этом повторный припадок в половине случаев происходит в течение ближайшего полугода [3,18,28,34,64,80]. В этих случаях оправдано и предпочтительно отсроченное лечение [64, 80]. У больных с неврологическим дефицитом и/или патологическими изменениями на ЭЭГ риск повторного генерализованного тонико–клонического припадка варьирует от 36 до 77% [27,28,64,80]. Решение о немедленном лечении принимается индивидуально в каждом случае. Риск повторения после двух припадков, по данным различных авторов, приближается к 80–90%. Данные пациенты должны лечиться АЭП [35,37,80].
Вид эпилепсии и АЭП
Выбор АЭП определяется видом эпилепсии. Наиболее частым (70%) видом эпилепсий взрослых являются локализационно–обусловленные (парциальные) эпилепсии [40,49], которые характеризуются простыми парциальными, комплексными парциальными и вторично генерализованными тонико–клоническими припадками. Чаще всего у взрослых наблюдаются комплексные парциальные припадки [30,36,75], причем их удельный вес с возрастом увеличивается [36]. Более чем у половины всех пациентов с парциальными эпилепсиями имеются как парциальные, так и вторично генерализованные тонико–клонические припадки и, следовательно, они нуждаются в АЭП, которые воздействуют на оба типа припадков [47,49]. Хотя генерализованные эпилепсии обычно начинаются в детстве, часть их не диагностируется до присоединения тонико–клонических припадков после второго десятилетия жизни [49]. Препаратами первой линии выбора в лечении парциальных эпилепсий остаются карбамазепин и фенитоин [19,49]. Несколько менее эффективными являются фенобарбитал и вальпроат [46,47,61,76]. Многочисленными исследованиями показано, что монотерапия с одним из этих стандартных АЭП обеспечивает адекватный контроль примерно у 70% взрослых пациентов с парциальной эпилепсией [48]. Еще у 15–20% пациентов ЭП прекращаются при использовании политерапии. У 10–15% пациентов припадки остаются рефрактерными к лечению стандартными АЭП, в связи с чем именно эти больные становятся кандидатами для терапии новыми препаратами или хирургического лечения [49].
Появление новых АЭП требует определения их места в лечении больных с парциальными эпилепсиями. Поскольку эффективность и безопасность новых АЭП окончательно не определены, их рекомендуют использовать при недостаточной контролируемости припадков или при выраженных побочных действиях от приема стандартных АЭП [49]. Исследования последних лет показали значительно большую эффективность монотерапии фелбаматом, габапентином, вигабатрином и ламотриджином по сравнению с плацебо и в качестве дополнительной терапии у пациентов с парциальными и вторично генерализованными тонико–клоническими припадками. Терапия приводила к более чем 50% уменьшению числа припадков у 25–30% [48], а по данным последних исследований у 54, 57,9 и 38% пациентов с рефрактерными припадками, принимавших вигабатрин, ламотриджин и габапентин соответственно [23,50,81]. Монотерапия ламотриджином у пациентов с впервые диагностированной эпилепсией имела такую же эффективность, как и лечение карбамазепином, но несколько лучше переносилась [6,9,45,72]. По сравнению с вальпроатом ламотриджин был более эффективным [31]. Исследованиями Brodie M.J и Pellok J.M. (1995) была показана эффективность и монотерапии фелбаматом, однако редкие осложнения в виде апластической анемии и печеночной недостаточности заставляют крайне осторожно подходить к назначению этого препарата. У больных с рефрактерными припадками использование топирамата в качестве монотерапии было эффективно у 45–46% [15,66], габапентина – у 54% больных с простыми парциальными, у 43% с комплексными парциальными и у 13% с вторично генерализованными припадками [5]. Тиагабин был эффективен у 53% пациентов с впервые диагностированными припадками [38,63]. Клинические исследования тиагабина, топирамата и вигабатрина показали, что они имеют эффективность, сравнимую или несколько большую, чем габапентин и ламотриджин [44].
Препаратом первой линии выбора при лечении первично генерализованных эпилепсий является вальпроат [19,49,64,67]. Он обеспечивает полный или частичный контроль припадков у 70–90% пациентов [16,69]. Вальпроат и ламотриджин обладают синергизмом в предотвращении первично генерализованных припадков [10]. Фенитоин и карбамазепин также могут быть эффективны при первично генерализованных тонико–клонических припадках, хотя ухудшают течение эпилепсий с абсансными и миоклоническими припадками [64].
В таблице 1 приведены данные по эффективности АЭП при различных видах припадков и механизмы действия АЭП.

Основными принципами фармакотерапии эпилепсий являются: 1) использование по возможности монотерапии; 2) начало лечения с низких доз для обеспечения среднего или более низкого уровня сывороточной концентрации препарата; 3) при необходимости увеличение дозы препарата, которое проводится только после достижения его стационарной (постоянной) концентрации в сыворотке, то есть не ранее чем через пять периодов полужизни АЭП. Увеличение дозы продолжается до прекращения припадков или появления побочных эффектов; 4) прием АЭП с частотой, равной периоду его полужизни и периоду полужизни его биологической активности; 5) переход на монотерапию другим АЭП или комбинированное лечение двумя АЭП при неэффективности монотерапии; 6) учет возможности межлекарственного взаимодействия; 7) контроль сывороточной концентрации АЭП при подозрении на токсическое действие, изменение фармакокинетики или несоблюдение режима приема АЭП; 8) при контролируемости припадков и отсутствии побочных эффектов дозы АЭП должны оставаться неизменными независимо от общепринятых доз и сывороточной концентрации АЭП.
Основными принципами фармакотерапии эпилепсий являются: 1) использование по возможности монотерапии; 2) начало лечения с низких доз для обеспечения среднего или более низкого уровня сывороточной концентрации препарата; 3) при необходимости увеличение дозы препарата, которое проводится только после достижения его стационарной (постоянной) концентрации в сыворотке, то есть не ранее чем через пять периодов полужизни АЭП. Увеличение дозы продолжается до прекращения припадков или появления побочных эффектов; 4) прием АЭП с частотой, равной периоду его полужизни и периоду полужизни его биологической активности; 5) переход на монотерапию другим АЭП или комбинированное лечение двумя АЭП при неэффективности монотерапии; 6) учет возможности межлекарственного взаимодействия; 7) контроль сывороточной концентрации АЭП при подозрении на токсическое действие, изменение фармакокинетики или несоблюдение режима приема АЭП; 8) при контролируемости припадков и отсутствии побочных эффектов дозы АЭП должны оставаться неизменными независимо от общепринятых доз и сывороточной концентрации АЭП.Предпочтительность монотерапии определяется различиями в метаболизме АЭП, что создает возможности межлекарственного взаимодействия и усиления побочных эффектов друг друга. Большинство АЭП выводятся из организма путем преобразования ферментами печени в соединения, которые выделяются почками. Метаболизм АЭП происходит по одному из трех типов кинетик фермента: дозонезависимой (линейной), дозозависимой (нелинейной) и время–зависимой кинетике.
Метаболизм большинства стандартных и более новых АЭП, за исключением карбамазепина, фенитоина и вальпроата, подчиняется дозонезависимой кинетике. Для нее характерно отсутствие влияния сывороточной концентрации на клиренс препарата и период его полужизни. Стационарная сывороточная концентрация имеет линейную зависимость от дозы применяемого АЭП. После прекращения приема АЭП с линейной кинетикой его концентрация в сыворотке уменьшается более чем на 95% после пяти периодов полужизни. И, наоборот, пять периодов полужизни необходимы для достижения стационарной сывороточной концентрации после начала лечения АЭП, после чего можно судить об эффективности принимаемой дозы и повышать ее в случае необходимости.
При дозозависимой (нелинейной) кинетике стационарная сывороточная концентрация препарата изменяется по нелинейной форме по отношению к дозе: сывороточная концентрация нарастает быстрее, чем увеличиваемая доза, и уменьшается быстрее, чем снижаемая доза. Клиренс лекарственного средства изменяется обратно пропорционально его сывороточной концентрации, а период полужизни лекарственного средства изменяется непосредственно с изменяемой сывороточной концентрацией. Данный тип метаболизма обусловлен насыщением метаболизирующего фермента субстратом, что приводит к уменьшению клиренса АЭП и, следовательно, к дополнительному повышению сывороточной концентрации и снижению лекарственного клиренса. Этот цикл самораспространения может потребовать длительного времени для своего завершения, т.e. для достижения стационарной концентрации [77]. Нелинейной кинетикой обладает фенитоин. Хотя фенитоин имеет период полужизни примерно 1 день, может потребоваться до 28 дней, чтобы достичь стационарной сывороточной концентрации после того, как доза была изменена [12,77]. В условиях удлиненного периода полужизни препарата его нарастающая сывороточная концентрация приводит к быстрому появлению у пациента признаков интоксикации [12]. Поэтому вне зависимости от массы тела рекомендуется начинать прием фенитоина с 300 мг в день, что у большинства больных обеспечивает минимальную сывороточную концентрацию.
При время–зависимой кинетике АЭП активируют ферменты, осуществляющие его метаболизм. Через несколько недель от начала лечения без изменения дозы данные процессы аутоиндукции приводят к увеличению клиренса АЭП, а следовательно, и к укорочению периода его полужизни и уменьшению сывороточной концентрации АЭП. Это делает необходимым увеличение дозы применяемого АЭП. Данный тип кинетики характерен для карбамазеина.
Целесообразность начала лечения с низких доз обусловлена индивидуальными особенностями метаболизма АЭП у каждого пациента. В большинстве случаев эффективное лечение коррелирует с рекомендуемыми диапазонами сывороточной концентрации. Однако в некоторых случаях припадки прекращаются при уровне сывороточной концентрации ниже терапевтического, в других – выше терапевтического уровня, при этом у пациента отсутствуют признаки интоксикации. Наконец, есть пациенты, у которых прием АЭП сопряжен с выраженной интоксикацией при уровне сывороточной концентрации ниже терапевтического диапазона [70]. Большинство исследователей рекомендуют начинать прием в дозе не более 1/3 от предполагаемой поддерживающей, что обеспечивает средний или более низкий уровень сывороточной концентрации.
Частота приема большинства АЭП зависит от периода его полужизни (время, в течение которого сывороточная концентрация уменьшается наполовину) и периода полужизни его биологической активности (время, в течение которого фармакологическое действие уменьшается наполовину) [14]. Для большинства стандартных АЭП период полужизни больше или равен периоду полужизни его биологической активности. AЭП с периодом полужизни 24 часа и более обычно даются только один раз в день. Рекомендуется принимать их перед сном, для избежания седативного действия, сопутствующего пиковому уровню АЭП. Лекарства с периодом полужизни менее 24 часов обычно даются с интервалом, приблизительно равным одному периоду полужизни. Увеличение дозы применяемого АЭП может удлинить время, в течение которого сывороточные концентрации остаются в терапевтическом диапазоне. Однако это приводит к большим колебаниям сывороточной концентрации препарата, увеличивает риск его токсичности на пике концентрации, а также повышает риск развития припадка при низкой концентрации.
Принципы классической сывороточной фармакокинетики не могут быть использованы для характеристики биологической активности АЭП, у которых период полужизни биологической активности значительно превышает период полужизни вследствие устойчивого связывания АЭП с высокоаффинными участками в мозге (например, каналами натрия, NMDA рецепторами, ГАМК–трансаминазой и др.). Примером АЭП с таким профилем является вигабатрин [41]. Его период полужизни составляет 5–7 часов, а период полужизни биологической активности – от 1 до нескольких дней, вследствие необратимой инактивации ГАМК–трансаминазы [55]. В данном случае рекомендуется одно–, двухкратный прием препарата в течение дня.
При неэффективности монотерапии одним из АЭП используют комбинированное лечение двумя АЭП, что создает возможности фаpмакодинамических и фармакокинетических лекарственных взаимодействий. Фармакодинамические взаимодействия происходят, когда два и более лекарства действуют в общем рецепторном месте. Взаимодействие может быть агонистическим или антагонистическим. Например, ламотриджин усиливает токсическое действие карбамазепина без изменения его сывороточной концентрации [53]. Фармакокинетические взаимодействия характерны для АЭП, метаболизирующихся ферментами печени [11]. В этом случае АЭП, подвергающийся метаболизму одним ферментом, может индуцировать или ингибировать фермент, осуществляющий метаболизм другого. Эти взаимодействия могут быть и двунаправленными (на каждое лекарственное средство может влиять фармакокинетика другого). Так, например, при использовании комбинации карбамазепина и фенитоина, карбамазепин ингибирует метаболизм фенитоина, а фенитоин индуцирует метаболизм карбамазепина, что приводит к повышению сывороточной концентрации фенитоина и снижению – карбамазепина [13,58]. В других случаях карбамазепин, фенитоин, фенобарбитал, почти всегда выступают мощными индукторами ферментов печени, что увеличивает клиренс одновременно применяемых препаратов и, следовательно, снижает их плазменную концентрацию. Среди более новых АЭП габапентин, вигабатрин и, в меньшей степени, ламотриджин не подвержены межлекарственным взаимодействиям. Каждый раз добавление нового лекарственного средства пациенту, принимающему АЭП, требует от лечащего врача согласования с перечнем возможных взаимодействий лекарственных средств [73]. Другим видом фармакокинетического взаимодействия является замещение из протеиносвязывающих участков одного АЭП другим [11]. Так, например, вальпроат и фенитоин, обладающие высокой способностью связывания с белками, могут быть замещены другими одновременно принимаемыми препаратами. Это приводит к временному увеличению уровня свободного препарата. Сходная ситуация наблюдается при снижении возможности связывания с белками, например, при гипоальбуминемии вследствие недостаточности почек, печени, при обширных ожогах, беременности и др. Поскольку именно свободная (несвязанная) фракция препарата проникает в мозг и приводит к лечебному действию, повышение дозы в данных условиях для поддержания терапевтического уровня сывороточной концентрации может привести к выраженным проявлениям интоксикации [1,49,64].
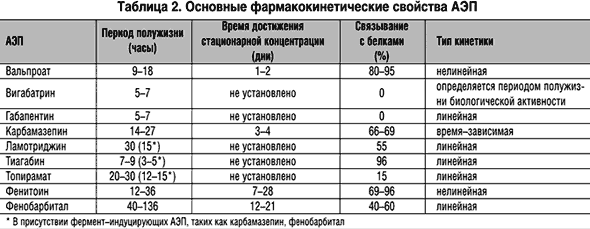
В таблице 2 приведены основные фармакокинетические свойства наиболее часто применяемых АЭП [14].
Фенитоин. Наиболее частая форма выпуска – 50 и 100 мг/табл. Начальная доза для взрослых – 300 мг 1 раз в сутки вне зависимости от массы тела. При продолжении припадков разовая доза увеличивается на 25–30 мг. Наиболее частой клинической ошибкой является увеличение дозы препарата с 300 мг/сут до 400 мг/сут, что приводит к развитию побочных действий через различные промежутки времени [1]. Межлекарственные взаимодействия обусловлены: 1) вытеснением фенитоина из связанного с белками плазмы состояния вальпроатом, гипогликемическими препаратами, ацетилсалициловой кислотой, варфарином, трициклическими антидепрессантами; 2) способностью фенитоина индуцировать ферменты печени, ускоряя метаболизм других АЭП, антикоагулянтов непрямого действия, стероидов, тироксина, трициклических антидепрессантов, доксициклина, фолатов. Метаболизм фенитоина ингибируется под влиянием циметидина, ко-тримоксазола, изониазида, левомицетина. В начале лечения возможны побочные реакции в виде сонливости, снижения внимания, двоения, пошатывания, что делает необходимым снижения дозы. У большинства пациентов в той или иной степени наблюдается гиперплазия десен и избыточное оволосение. Длительное применение препарата может привести к периферической нейропатии и остеомаляции [1, 19].
. Наиболее частая форма выпуска – 50 и 100 мг/табл. Начальная доза для взрослых – 300 мг 1 раз в сутки вне зависимости от массы тела. При продолжении припадков разовая доза увеличивается на 25–30 мг. Наиболее частой клинической ошибкой является увеличение дозы препарата с 300 мг/сут до 400 мг/сут, что приводит к развитию побочных действий через различные промежутки времени [1]. Межлекарственные взаимодействия обусловлены: 1) вытеснением фенитоина из связанного с белками плазмы состояния вальпроатом, гипогликемическими препаратами, ацетилсалициловой кислотой, варфарином, трициклическими антидепрессантами; 2) способностью фенитоина индуцировать ферменты печени, ускоряя метаболизм других АЭП, антикоагулянтов непрямого действия, стероидов, тироксина, трициклических антидепрессантов, доксициклина, фолатов. Метаболизм фенитоина ингибируется под влиянием циметидина, ко-тримоксазола, изониазида, левомицетина. В начале лечения возможны побочные реакции в виде сонливости, снижения внимания, двоения, пошатывания, что делает необходимым снижения дозы. У большинства пациентов в той или иной степени наблюдается гиперплазия десен и избыточное оволосение. Длительное применение препарата может привести к периферической нейропатии и остеомаляции [1, 19].Карбамазепин. Наиболее частая форма выпуска 200 мг/табл. Начальная доза составляет не более 1/3 от предполагаемой поддерживающей. Поддерживающая доза составляет 400–1800 мг/сутки. Рекомендуется двухкратный прием в течение дня. Межлекарственные взаимодействия обусловлены индуцированием активности ферментов печени, принимающих участие в метаболизме фенитоина, вальпроата, варфарина, стероидов, оральных контрацептивов, что приводит к снижению их концентрации в сыворотке [19]. С другой стороны, фенобарбитал и фенитоин, также являясь мощными индукторами печеночных ферментов, снижают уровень карбамазепина [19]. Примерно у трети больных, получающих карбамазепин, отмечаются побочные эффекты, обычно не требующие отмены препарата: сонливость, диплопия, атаксия, неприятные ощущения в желудке, эритематозная кожная сыпь [19].
Вальпроат. Наиболее частая форма выпуска – 200, 300 и 500 мг/табл. Начальная доза составляет 400–600 мг (10 мкг/кг), поддерживающая 600–3000 (20–60 мкг/кг). Рекомендуется двух–, трехкратный прием в течение дня. Поскольку 90% вальпроата связано с белками сыворотки, то значительное повышение дозы, из–за насыщения протеин–связывающих участков, приводит к увеличению свободной фракции препарата и появлению токсических эффектов, выраженность которых может варьировать из–за колебаний уровня свободной фракции [64]. Межлекарственные взаимодействия с другими препаратами обусловлены ингибированием активности ферментов печени. При одновременном приеме вальпроат повышает плазменные концентрации фенитоина, фенобарбитала, карбамазепина и ламотриджина [1]. Начальная доза последнего при использовании с вальпроатом составляет обычно 0,2 мг/кг и может увеличиваться не чаще, чем через 2 недели до 100–150 мг/сутки [49,64]. Вальпроат может вытеснять фенитоин из его связей с сывороточными белками [1]. Фелбамат ингибирует метаболизм вальпроата, что приводит к значительному повышению его плазменной концентрации и требует уменьшения дозы на 30–50% [64]. Побочные действия отмечаются, как правило, при назначении больших доз препарата и включают: желудочно–кишечный дискомфорт, сонливость, легкий тремор, иногда – повышенный аппетит и выпадение волос. Временное снижение дозы не только устраняет выраженность побочных эффектов, но и позволяет добиться более легкой переносимости высоких доз в дальнейшем [19]. Отмечена корреляция терапевтического уровня с тремором [62]. Идиосинкратическая токсичность вальпроата проявляется гепатотоксичностью различной степени тяжести, тромбоцитопенией и тератогенным действием. Риск развития осложнений возрастает при использовании политерапии [1].
Ламотриджин. Наиболее частая форма выпуска – 25, 50, 100, 200 мг/табл. Начальная доза составляет 25 мг/cут при использовании в качестве монотерапии (0,5 мг/кг); 12,5 мг/cут (0,2 мг/кг) при использовании с вальпроатом; 50 мг/сут (1,0 мг/кг), если используется с фермент–индуцирующими АЭП [53, 54]. Рекомендуется одно–, двухкратный прием в течение дня. Следует увеличивать дозу не чаще, чем с двухнедельным интервалом, в течение 8–12 недель [49]. Поддерживающая суточная доза составляет 100–150 мг при использовании с вальпроатом, 400–500 мг при одновременном назначении с фенитоином и карбамазепином [64]. Ламотриджин хорошо переносится. Наиболее часто встречаемым побочным действием является сыпь, которая наблюдается у 10% пациентов [63]. При одновременном использовании с карбамазепином увеличивается риск развития головокружения и диплопии, с фенитоином – атаксии, вальпроатом – тремора и сыпи [10]. Начало терапии с низких доз и медленное ее повышение снижают риск их развития [10,63].
Вигабатрин. Наиболее частая форма выпуска 500 мг/таб. Начальная доза препарата – 500 мг/сут, поддерживающая – 1500–2000 мг/сут. Рекомендуется двухкратный прием в течение дня [1,49,53]. Вигабатрин хорошо переносится, не снижает когнитивные функции или даже несколько улучшает их [56]. Однако у некоторых пациентов наблюдалось увеличение веса, сонливость, редко – психомоторное возбуждение, психоз, гипертрофический гингивит [26,39,78].
Габапентин. Наиболее частая форма выпуска – 300 и 400 мг/капс, 600 и 800 мг/табл. Начальная доза препарата 900 мг/сут, поддерживающая – 1800–4800 г/сут [5]. Рекомендуется его трехкратный прием в течение дня. Габапентин хорошо переносится большинством пациентов, хотя на начальном этапе лечения могут отмечаться тремор, атаксия, диплопия. Препарат не влияет на когнитивные функции при приеме в дозе 1200–2400 мг/сут [41]. При приеме более высоких доз отмечено появление сонливости [41]. Достоинством препарата является отсутствие межлекарственных взаимодействий.
Топирамат. Наиболее частая форма выпуска – 25, 100, 200 мг/табл. Начальная доза препарата – 12,5–25–50 мг/сут, поддерживающая 400–1000 мг/сут. Прием дозы свыше 1600 мг/cут не исследовался [49,65]. Рекомендуется двухкратный прием в течение дня [63,66]. Считается целесообразным увеличение дозы на 50 мг в неделю. На начальном этапе лечения могут отмечаться тремор, атаксия, диплопия, потеря веса [4,60,63,65].
Тиагабин. Рекомендуемая поддерживающая доза 15–30 мг/сут в 2–3 приема [38,63]. При использовании c фермент–индуцирующими препаратами может потребоваться увеличение дозы [63]. На начальном этапе лечения могут отмечаться тремор, атаксия, диплопия. Негативного влияния тиагабина на когнитивные функции и качество жизни не отмечено [24,25].
Прекращение лечения возможно при отсутствии припадков в течение не менее чем 2 лет для пациентов с низким и 3–5 лет – высоким риском рецидива припадков. Риск их возобновления определяется, в первую очередь, видом эпилепсии или эпилептического синдрома [19,64]. Так, например, ювенильная миоклоническая эпилепсия относится к рециди вирующим эпилепсиям и поэтому, несмотря на хорошую контролируемость припадков у большинства пациентов, прекращение лечения нежелательно. Имеется связь риска рецидива припадков с их типом: он минимален при типичных абсансах и последовательно повышается при следующих типах припадков: миоклонические, клонические, тонические, простые и комплексные парциальные припадки [2,19]. Наибольшая частота возобновления припадков отмечается у пациентов, которым длительно не удавалось подобрать эффективное лечение, принимавших различные препараты или их комбинации, а также при наличии значительных патологических изменений на ЭЭГ [51,52,57,64]. Благоприятный прогноз при отмене АЭП отмечен у пациентов с редкими припадками, эффективно контролируемыми монотерапией, нормальными результатами ЭЭГ [64]. На решение об отмене препарата влияет также выраженность у пациента токсических проявлений вследствие длительного приема АЭП [64]. Большинством исследователей рекомендуется медленное уменьшение дозы АЭП за 3–6 месяцев [19,64].
Фармакорезистентные припадки
Эпилептические припадки, продолжающиеся после двухлетнего лечения стандартными и более новыми АЭП и нарушающие нормальную жизнь пациента или его семьи, считаются фармакорезистентными (рефрактерными) [7]. Наиболее характерными предикторами рефрактерности припадков являются: ранний возраст их начала, частые генерализованные припадки, органическое или структурное поражение мозга в качестве причины припадков, низкий интеллект, наличие атонических и атипичных абсансных припадков, грубые изменения на ЭЭГ по эпилептическому и общепатологическому типу [7]. У большинства пациентов с поздним началом эпилепсии и невозможностью контроля припадков в течение двух лет лечения необходимо исключить причины псевдофармакорезистентности: неправильно поставленный диагноз и неадекватное лечение. При наличии истинной фармакорезистентности целесообразно рассмотреть вопрос о возможном хирургическом лечении. Основными критериями отбора пациентов на хирургическое лечение методом локализованной резекции мозга являются: операбельность структурного поражения, идентифицированного на МРТ; соответствие припадков очаговому поражению мозга (используя видеотелеметрию); отсутствие грубых нарушений психики; низкий коэффициент интеллекта; отсутствие прогрессирующих церебральных и системных заболеваний; отсутствие общих медицинских противопоказаний к операции [7,21,29].
Расширение спектра современных АЭП, изучение фармакокинетики и фармакодинамики их взаимоотношений является основой для успешного консервативного лечения эпилепсии.
1. Бертрам Г. Катцунг. Базисная и клиническая фармакология. Пер. с англ. – М. – СПб: Бином–Невский диалект, 1998.
2. Annegers JF, Hauser WA, Elveback LR. Remission of seizures and relapse in patients with epilepsy. Epilepsia 1979; 20:729–737.
3. Annegers JF, Shirts SB, Hauser WA, Kurland LT. Risk of recurrence after initial unprovoked seizure. Epilepsia 1986; 27: 43–50.
4. Ben–Menachem E, Dam M., Mikkelsen M et al. Topiramate add–on treatment in patients with intractable partial epilepsy: a multicentre study. Epilepsia 1993: 34 (suppl 2): 109.
5. Beydoun A, Fakhoury T, Nasreddine W, Abou–Khalil B. Conversion to high dose gabapentin monotherapy in patients with medically refractory partial epilepsy. Epilepsia 1998; 39:188–193.
6. Beydoun A. Monotherapy trials of new antiepileptic drugs. Epilepsia 1997; 38 (suppl 9): S21–S31.
7. Bourgeois BFD General concepts of medical intractability. In: Luders HO. Epilepsy surgery. New York: Raven Press, 1992: 77–82.
8. Brodie MJ, Pellock JM. Taming the brainstorms: felbamate updated. Lancet 1995; 346: 918–919.
9. Brodie MJ, Richens A, Yuen AWC et al. Double–blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. Lancet 1995; 345: 476–479.
10. Brodie MJ, Yuen AW. Lamotrigine substitution study: evidence for synergism with sodium valproate? 105 Study Group. Epilepsy Res 1997; 26: 423–432.
11. Browne TR, Greenblatt DJ, Schumacher GE et al. Comparison of methods for determination of pharmacokinetic drug interactions and proposals for new methods. In: Pitlick WH, ed. Antiepileptic drug interactions. New York: Demos, 1989: 1– 26.
12. Browne TR, LeDuc B. Phenytoin: biotransformation. In: Levy RH, Mattson RH, Meldrum BS, eds. Antiepileptic drugs. New York: Raven Press, 1995: 283–300.
13. Browne TR, Szabo GK, Evans JE et al. Carbamazepine increases phenytoin serum concentration and reduces phenytoin clearance. Neurology 1988; 38: 1146–1150.
14. Browne TR. Clinical pharmacology and phannacokinetics of antiepileptic drugs. In: Chokroverty S, ed. Modern management of epilepsy. New York: Butterworth, 1996: 47–65.
15. Bruni J. Efficacy of topiramate. Can J Neurol Sci 1998; 25: S6–S7.
16. Chadwick DW. Valproate monotherapy in the management of generalized and partial seizures. Epilepsia 1987; 28 (suppl): S12–S17.
17. Cheung H, Kamp D, Harris E. An in vitro investigation of the action of lamotrigine on neuronal voltage–activated sodium channels. Epilepsy Res 1992; 13: 89–92.
18. Cockerell OC, Hart YM, Sander JWAS, Shorvon SD. Pattern of seizure recurrence of newly diagnosed epilepsies in the National General Practice Experience. Epilepsia 1993; 34 (suppl 6): 138.
19. Cockerell OC, Shorvon SD. Epilepsy. Current Concepts. London: Current Medical Literature Ltd., 1996: 55–67.
20. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 1981;22:498–501.
21. Dasheiff RM, McNamara D, Dickinson LV Efficacyof second–line antiepileptic drugs in the treatment of patients with medically refractive complex partial seizures. Epilepsia 1986; 26: 124–127.
22. Davies J, Richens A. Neuropharmacology. In: Laidlaw J, Richens A, Chadwick D eds. A textbook of epilepsy. London: Churchill–Livingstone 1993: 495–559.
23. Dean C, Mosier M, Penry K. Dose–response study of vigabatrin as add–on therapy in patients with uncontrolled complex partial seizures. Epilepsia 1999 Jan; 40 (1): 74–82
24. Dodrill CB, Arnett JL, Shu V et al. Effects of tiagabine monotherapy on abilities, adjustment, and mood. Epilepsia 1998; 39: 33–42
25. Dodrill CB, Arnett JL, Sommerville KW, Shu V. Cognitive and quality of life effects of differing dosages of tiagabine in epilepsy. Neurology 1997; 48: 1025–1031.
26. Dodrill CB, Arnett JL, Sommerville KW, Sussman NM. Evalution of the effects of vigabatrin on cognitive abilities and quality of life in epilepsy. Neurology 1993; 43:2501–2507.
27. Elwes RD, Chesterman P, Reynolds EH. Prognosis after a first untreated tonic–clonic seizure. Lancet 1985; 2: 752–753.
28. Elwes RD, Johnson AL, Reynolds EH. The course of untreated epilepsy. Br Med J 1988; 297: 948–950.
29. Engel J, Shewmon DA. Who should be considered a surgical candidate? In: Engel J, ed Surgical treatment of the epilepsies. New York: Raven Press, 1993: 23–34.
30. Gastaut H., Gastaut J.L. et al. Relative frequency of different types of epilepsy: a stady employing the classification of the International League Agaist Epilepsy. Epilepsia 1975; 16: 457–461.
31. Gilliam F, Vazquez B, Sackellares JC. et al. An active–control trial of lamotrigine monotherapy for partial seizures. Neurology 1998; 51: 1018–1025.
32. Goodridge DM, Shorvon SD. Epileptic seizures in a population of 6000. Treatment and prognosis. Br. Med. J 1983; 287: 645–647.
33. Grimes DA. Unplanned pregnancies in the US. Obstet Gynecol 1986; 67: 438–442.
34. Hart YM, Sander JW, Johnson AL, Shorvon SD. National General Practice Study of Epilepsy: recurrence after a first seizure. Lancet 1990; 336: 1271–1274.
35. Hauser WA, Anderson VE, Loewenson RB, McRoberts SM. Seizure recurrence after a first unprovoked seizure. N Engl J Med 1982; 307: 522–528.
36. Hauser WA, Annegers JF, Kurland LT. The incidence of epilepsy in Rochester, Minnesota, 1935–1984. Epilepsia 1993; 34: 453–468.
37. Hauser WA, Rich SS, Lee JR, Annegers JF, Anderson VE. Rise of recurrent seizures after two unprovoked seizures. N Engl J Med 1998; 338: 429–434.
38. Herranz JL. Tiagabine: recent findings and recommendations for dosage. Rev Neurol 1998; 27: 101–103.
39. Katz J, Givol N, Chaushu G et al. Vigabatrin–induced gingival overgrowth. J Clin Periodontol 1997; 24: 180–182.
40. Keranen T, Sillanpaa M, Riekkinen PJ. Distribution of seizure types in an epileptic population. Epilepsia 1988; 29: l–7.
41. Leach JP, Girvan J, Paul A, Brodie MJ. Gabapentin and cognition: a double blind, dose ranging, placebo controlled study in refractory epilepsy. J Neurol Neurosurg Psychiatry 1997; 62: 372–376.
42. Lewis PJ, Richens E. Vigabatrin: a new antiepileptic drug. Br J Clin Pharmacol 1989; 27 (suppl 1): 1S–12S.
43. Macdonald RL, Meldrum BS. Principles of antiepileptic drug action. In: Antiepileptic drugs, 3rd ed. Levy RH et al. (eds) Raven Press 1989.
44. Marson AG, Kadir ZA, Chadwick DW. New antiepileptic drugs: a systemic review of their efficacy and tolerability. BMJ 313; 1996: 1169–1174.
45. Matsuo F, Bergen D, Faught E et al. Placebo–controlled study of the efficacy and safety of lamotrigine in patients with partial seizures. Neurology 1993; 43: 2284–2291.
46. Mattson RH, Cramer JA, Collins JF et al. A comparison of valproate with carbamazepine for the treatment of complex partial seizures and secondarily generalized tonic–clonic seizures in adults. N Engl J Med 1992; 327: 765–771.
47. Mattson RH, Cramer JA, Collins JF et al. Comparison of carbamazepine, phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic–clonic seizures. N Engl J Med 1985; 313: 145–151.
48. Mattson RH, Cramer JA. The choice of antiepileptic drugs in focal epilepsy. In: Wylie E, ed. The treatment of epilepsy: principles and practices. Philadelphia: Lea & Febiger, 1993: 817–823.
49. Mattson RH. Medical management of epilepsy in adults. Neurology 1998; 51 (suppl 4): S15–S20.
50. Mayer T, Schutte W, Wolf P, Elger CE. Gabapentin add–on treatment: how many patients become seizure–free? An open–label multicenter study. Acta Neurol Scand 1999; 99: 1–7.
51. Medical Research Council Antiepileptic Drug Withdrawal Study Group. Prognostic index for recurrence of seizures after remission of epilepsy. Br Med J 1993; 306: 1374–1378.
52. Medical Research Council Antiepileptic Drug Withdrawal Study Group. Randomised study of antiepileptic drug withdrawal in patients in remission. Lancet 1991; 337: 1175–1180.
53. Messenheimer JA. Lamotrigine. Clin Neuropharmacol 1994; 17: 548–559.
54. Messenheimer JA. Lamotrigine. Epilepsia 1995; 36 (suppl 2): S87–S94.
55. Messenheimer JA. Vigabatrin. Clin Neuropharmacol 1994; 17: 548–559.
56. Monaco F, Torta R, Cicolin A et al. Lack of association between vigabatrin and impaired cognition. J Int Med Res 1997; 25: 296–301.
57. Overweg J, Binnie CD, Oosting J, Rowan AJ. Clinical and EEG prediction of seizure recurrence following antiepileptic drug withdrawal. Epilepsy Res 1987; 1: 272–283.
58. Perucca E. Pharmacokinetic interactions with antiepileptic drugs. Clin Pharmacokinet 1982; 7: 57–84.
59. Pierce MW, Suzdack PD, Gustavason LE et al. New antiepileptic drugs. Epilepsy Res 1991; suppl 3.
60. Privitera MD. Topiramate: a new antiepileptic drug. Ann Pharmacother 1997; 31: 1164–1173.
61. Ramsay RE, Wilder BJ, Berger JR et al. A double–blind study comparing carbamazepine with phenytoin as initial seizure therapy in adults. Neurology 1983; 33: 904–910.
62. Richens A, Perucca E. Clinical pharmacology and medical treatment. In: Laidlaw J, Richens A, Chadwick D eds. A textbook of epilepsy. London: Churchill–Livingstone 1993: 495–559.
63. Rowan AJ. Reflections on the treatment of seizures in the elderly population. Neurology 1998; 51 (suppl 4): S28–S33.
64. Rowland LP Merritt’s textbook of neurology. Williams& Wilkins 1995: 845–869.
65. Sachdeo RC, Reife RA, Lim P, Pledger G. Topiramate monotherapy for partial onset seizures. Epilepsia 1997; 38: 294–300.
66. Sachdeo RC. Topiramate. Clinical profile in epilepsy. Clin Pharmacokinet 1998; 34: 335–346.
67. Sander JW, Hart YM, Johnson AL, Shorvon SD. National General Practice Study of Epilepsy: newly diagnosed epileptic seizures in a general population Lancet 1990; 336: 1267–1271.
68. Sander JW, Shorvon SD. Incidence and prevalence studies in epilepsy and their methodological problems: a review. J Neurol Neurosurg Psychiatry 1987; 50: 829–839.
69. Sato S, White BG, Penry JK, et al. Valproic acid versus ethosuximide in the treatment of absence seizures. Neurology 1982; 32: 157–163.
70. Schumacher GE, Barr JT, Browne TR, Collins JF, and the VA Epilepsy Cooperative Study Group. Test performance characteristics of the serum phenytoin concentration (SPC): the relationship between SPC and patient response. Ther Drug Monit 1991; 13: 318–324.
71. Steiner TJ. Comparison of lamotrigine and phenytoin monotherapy in newly diagnosed epilepsy [Abstract]. Epilepsia 1994; 35 (suppl 8): 31.
72. Stefani A, Spadoni F, Bernardi G. Gabapentin inhibits calcium currents in isolated rat brain neurons. Neuropharmacology 1998; 37: 83–91.
73. Tatro DS. Drug interaction facts. St. Louis: Facts and Comparison 1997.
74. Taylor CP, Gee NS, Su T, Kocsis JD et al. A summary mechanistic hypotheses of gabapentin pharmacology. Epilepsy Res 1998; 29: 233–249.
75. Theodore R.J., Porter R.J. Epilepsy. 100 Elementary Principles. 3th Edition. Saunders W.S. Company ltd. London 1995.
76. Treiman DM. Efficacy and safety of antiepileptic drugs: a review of controlled trials. Epilepsia 1987; 28 (suppl): SI–S8.
77. Wagner J. Time to reach steady state and prediction of steady state concentration for drugs obeying Michaelis–Menten elimination kinetics. J Phannacokinet Biopharm 1978; 6: 209–225.
78. Walker MC, Sander JWAS. Developments in antiepileptic drug therapy. Curr Opin Neurol Neorosurg 1994; 7: 131–139.
79. White HS, Brown SD, Woodhead JH et al. Topiramate enhances GABA–mediated chloride flux and GABA–evoked chloride currents in murine brain neurons and increases seizure threshold. Epilepsy Res 1997; 28: 167–179.
80. Willmore LJ. Epilepsy emergencies. The first seizure and status epilepticus. Neurology 1998; 51 (suppl 4): S34–S38.
81. Yen DJ, Yiu CH, Kwan SY et al. Lamotrigine as add–on therapy in adult patients with refractory epilepsy. Chung Hua I Hsueh Tsa Chih (Taipei) 1997; 59: 303–307.



