




–Р–≤—В–Њ—А—Л –њ—А–Є–≤–Њ–і—П—В –ї–Є—В–µ—А–∞—В—Г—А–љ—Л–µ –і–∞–љ–љ—Л–µ, —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—Й–Є–µ –Њ —В–Њ–Љ, —З—В–Њ –њ—А–Є –Я–Ю–£–У –≤ –њ—А–Њ—Ж–µ—Б—Б –≤–Њ–≤–ї–µ—З–µ–љ—Л –љ–µ —В–Њ–ї—М–Ї–Њ –≥–∞–љ–≥–ї–Є–Њ–љ–∞—А–љ—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞ —Б–µ—В—З–∞—В–Ї–Є –Є –і–Є—Б–Ї –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞, –љ–Њ –Є —Ж–µ–љ—В—А–∞–ї—М–љ—Л–µ –Њ—В–і–µ–ї—Л –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞—В–Њ—А–∞. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –љ–∞–њ—А–∞–≤–ї–µ–љ—Л –љ–∞ –Є–Ј—Г—З–µ–љ–Є–µ –њ—А–Є—З–Є–љ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –≥–ї–∞—Г–Ї–Њ–Љ–љ–Њ–є –љ–µ–є—А–Њ–Њ–њ—В–Є–Ї–Њ–њ–∞—В–Є–Є –Є —Б–љ–Є–ґ–µ–љ–Є—П –Ј—А–µ–љ–Є—П.
–Ъ–ї—О—З–µ–≤—Л–µ —Б–ї–Њ–≤–∞: –Я–Ю–£–У, –љ–µ–є—А–Њ–Њ–њ—В–Є–Ї–Њ–њ–∞—В–Є—П, –∞–љ—В–Є–Њ–Ї—Б–Є–і–∞–љ—В—Л.
Abstract
Neurodegenerative changes in the brain in glaucoma.
Literary review
I.R. Gazizova, Almaisam Rindzhibal
North–Western State Medical University named after
Mechnikov I.I. of the Ministry of health and Social Development of Russia, S.–Petersburg
Authors present literary data which gives a proof that in POAG process not only ganglian fibres and optic disc but also central parts of visual analyzer are involved.The main goal of the studies is an investigation of a reasons of the glaucoma neuroopticopathy and a decrease of a visual acuity.
Key words: POAG, neuroopticopathy, antioxidants.
–Х—Й–µ –≤ –Љ–Њ–љ–Њ–≥—А–∞—Д–Є–Є –†. –С–Є–љ–≥–∞ –Є –†. –С—А—Г–Ї–љ–µ—А–∞ –њ–Њ–і —А–µ–і–∞–Ї—Ж–Є–µ–є –Х.–Ц. –Ґ—А–Њ–љ–∞ «–Ь–Њ–Ј–≥ –Є –≥–ї–∞–Ј» (1959) —Г–њ–Њ–Љ–Є–љ–∞–µ—В—Б—П –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –љ–µ–Љ–µ—Ж–Ї–Є—Е —Г—З–µ–љ—Л—Е, –≥–і–µ –Њ–њ–Є—Б—Л–≤–∞–µ—В—Б—П «–Њ—А–≥–∞–љ–Є—З–µ—Б–Ї–∞—П –≥–Є–±–µ–ї—М –љ–µ—А–≤–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –≤ —В—А–µ—Е —Б–ї–Њ—П—Е –љ–∞—А—Г–ґ–љ–Њ–≥–Њ –Ї–Њ–ї–µ–љ—З–∞—В–Њ–≥–Њ —В–µ–ї–∞ —Г –Њ–±–µ–Ј—М—П–љ—Л –њ–Њ—Б–ї–µ —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ–Њ–≥–Њ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П —Б–µ—В—З–∞—В–Ї–Є» (—А–Є—Б. 1). –Т –њ–Њ—Б–ї–µ–і–љ–µ–µ –≤—А–µ–Љ—П –Љ–љ–Њ–≥–Є–µ –∞–≤—В–Њ—А—Л —Б—Е–Њ–і—П—В—Б—П –љ–∞ —В–Њ–Љ, —З—В–Њ –і–µ–≥–µ–љ–µ—А–∞—Ж–Є—П –љ–µ–є—А–Њ—Н–ї–µ–Љ–µ–љ—В–Њ–≤ –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞ –Љ–Њ–ґ–µ—В –њ—А–Њ–Є—Б—Е–Њ–і–Є—В—М –≤ –њ—А–Њ—Ж–µ—Б—Б–µ –≤—В–Њ—А–Є—З–љ–Њ–є —В—А–∞–љ—Б—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є (–і–µ–≥–µ–љ–µ—А–∞—Ж–Є—П –љ–µ—А–≤–љ—Л—Е –Ї–ї–µ—В–Њ–Ї, –Њ–±—А–∞–Ј—Г—О—Й–Є—Е –Њ–і–Є–љ —Б–Є–љ–∞–њ—Б, –њ—А–Є –љ–∞—А—Г—И–µ–љ–Є–Є –Є—Е –∞—Д—Д–µ—А–µ–љ—В–љ–Њ–є —Б–≤—П–Ј–Є), –њ—А–Є —Н—В–Њ–Љ –Ї–ї—О—З–µ–≤—Л–Љ —Н–ї–µ–Љ–µ–љ—В–Њ–Љ –µ–µ —А–∞–Ј–≤–Є—В–Є—П —П–≤–ї—П–µ—В—Б—П –∞–Ї—Б–Њ–љ–Њ–њ–∞—В–Є—П.
–Э–µ—Б–Љ–Њ—В—А—П –љ–∞ –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —Н—В–Є–Њ–ї–Њ–≥–Є–Є –Є –њ–∞—В–Њ–≥–µ–љ–µ–Ј–∞ –њ–µ—А–≤–Є—З–љ–Њ–є –Њ—В–Ї—А—Л—В–Њ—Г–≥–Њ–ї—М–љ–Њ–є –≥–ї–∞—Г–Ї–Њ–Љ—Л (–Я–Ю–£–У), —Г –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –±–Њ–ї—М–љ—Л—Е —Б –і–ї–Є—В–µ–ї—М–љ—Л–Љ —В–µ—З–µ–љ–Є–µ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –і–∞–ґ–µ –љ–∞ —Д–Њ–љ–µ –љ–Њ—А–Љ–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–≥–Њ –≤–љ—Г—В—А–Є–≥–ї–∞–Ј–љ–Њ–≥–Њ –і–∞–≤–ї–µ–љ–Є—П (–Т–У–Ф) –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ—А–Њ–≥—А–µ—Б—Б–Є–≤–љ–Њ–µ —Г—Е—Г–і—И–µ–љ–Є–µ –Ј—А–Є—В–µ–ї—М–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є —Б –њ–µ—А–µ—Е–Њ–і–Њ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –≤ –±–Њ–ї–µ–µ —В—П–ґ–µ–ї—Г—О —Б—В–∞–і–Є—О. –Ъ —Б–Њ–ґ–∞–ї–µ–љ–Є—О, –њ—А–Њ—Ж–µ—Б—Б –Є–Љ–µ–µ—В —Б—В—А–Њ–≥–Њ –Њ–і–љ–Њ–љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л–є –≤–µ–Ї—В–Њ—А —А–∞–Ј–≤–Є—В–Є—П, –њ—А–Є—З–µ–Љ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–є. –Э–∞—И –Њ–±–Ј–Њ—А –ї–Є—В–µ—А–∞—В—Г—А—Л –Њ–±–Њ–±—Й–∞–µ—В –і–∞–љ–љ—Л–µ –Њ –≤–µ—А–Њ—П—В–љ–Њ—Б—В–Є —В–Њ–≥–Њ, —З—В–Њ –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ—А–Є –Я–Ю–£–У –њ—А–Њ–Є—Б—Е–Њ–і—П—В –љ–µ —В–Њ–ї—М–Ї–Њ –≤ —Б–µ—В—З–∞—В–Ї–µ –Є –і–Є—Б–Ї–µ –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞, –љ–Њ –Є –љ–∞ –њ—А–Њ—В—П–ґ–µ–љ–Є–Є –≤—Б–µ–≥–Њ –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –њ—Г—В–Є.
–Ф–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, –њ—А–Њ–Є—Б—Е–Њ–і—П—Й–Є–µ –≤ –≥–Њ–ї–Њ–≤–љ–Њ–Љ –Љ–Њ–Ј–≥–µ, –і–µ—В–∞–ї—М–љ–Њ –Є–Ј—Г—З–∞—О—В—Б—П –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї—П–Љ–Є —В–∞–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –Ї–∞–Ї –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞ –Є –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞. –Э–µ–≤—А–Њ–ї–Њ–≥–Є –Њ–±–ї–∞–і–∞—О—В –љ–µ–Ї–Њ—В–Њ—А—Л–Љ –∞—А—Б–µ–љ–∞–ї–Њ–Љ —Б—А–µ–і—Б—В–≤ –і–ї—П –њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є—П –Є/–Є–ї–Є –Ј–∞–Љ–µ–і–ї–µ–љ–Є—П –љ–µ–є—А–Њ–љ–∞–ї—М–љ–Њ–є –≥–Є–±–µ–ї–Є [4]. –°—Г—Й–µ—Б—В–≤—Г—О—В —Г–±–µ–і–Є—В–µ–ї—М–љ—Л–µ –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М—Б—В–≤–∞ —В–Њ–≥–Њ, —З—В–Њ —Ж–µ–љ—В—А–∞–ї—М–љ–∞—П —А–Њ–ї—М –≤ –њ—А–Њ—Ж–µ—Б—Б–∞—Е –љ–µ–Њ–±—А–∞—В–Є–Љ–Њ–є –≥–Є–±–µ–ї–Є –љ–µ—А–≤–љ–Њ–є –Ї–ї–µ—В–Ї–Є –њ—А–Є–љ–∞–і–ї–µ–ґ–Є—В –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є—П–Љ [7,10]. –Ш –Є–Љ–µ–љ–љ–Њ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–Є —П–≤–ї—П—О—В—Б—П –Љ–Є—И–µ–љ—М—О –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –љ–µ–є—А–Њ–њ—А–Њ—В–µ–Ї—В–Њ—А–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ [11].
–У–ї–∞—Г–Ї–Њ–Љ–∞ –Ї–∞–Ї –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ. –Т –њ–Њ—Б–ї–µ–і–љ–Є–µ –≥–Њ–і—Л –Я–Ю–£–У –Њ—В–љ–Њ—Б—П—В –Ї –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П–Љ [22]. –Ю—Б–љ–Њ–≤–љ–Њ–є –њ—А–Є—З–Є–љ–Њ–є —А–∞–Ј–≤–Є—В–Є—П –љ–µ–є—А–Њ–Њ–њ—В–Є–Ї–Њ–њ–∞—В–Є–Є –њ—А–Є –≥–ї–∞—Г–Ї–Њ–Љ–µ –њ—А–Є–љ—П—В–Њ —Б—З–Є—В–∞—В—М –њ–Њ–≤—Л—И–µ–љ–љ–Њ–µ –Т–У–Ф. –Ч–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —Б –≤–Њ–Ј—А–∞—Б—В–Њ–Љ –Є —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –њ—А–Њ–≥—А–µ—Б—Б–Є–≤–љ—Л–Љ —В–µ—З–µ–љ–Є–µ–Љ, —Г –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –±–Њ–ї—М–љ—Л—Е – –і–∞–ґ–µ –љ–∞ —Д–Њ–љ–µ –љ–Њ—А–Љ–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–≥–Њ —Г—А–Њ–≤–љ—П –Њ—Д—В–∞–ї—М–Љ–Њ—В–Њ–љ—Г—Б–∞ [28]. –Ь–µ—Е–∞–љ–Є–Ј–Љ–Њ–Љ –≥–Є–±–µ–ї–Є –Ї–ї–µ—В–Њ–Ї —Б–µ—В—З–∞—В–Ї–Є –Є –∞–Ї—Б–Њ–љ–Њ–≤ –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞ –њ—А–Є –≥–ї–∞—Г–Ї–Њ–Љ–µ, –Ї–∞–Ї –Є –њ—А–Є –≤—Б–µ—Е –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞—Е, —П–≤–ї—П–µ—В—Б—П —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є –Ј–∞–њ—А–Њ–≥—А–∞–Љ–Љ–Є—А–Њ–≤–∞–љ–љ—Л–є –∞–њ–Њ–њ—В–Њ–Ј [31].
–Т 1996 –≥. –У.–Ш. –Ф–Њ–ї–ґ–Є—З —Б —Б–Њ–∞–≤—В. –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–ї, —З—В–Њ –њ—А–Њ—Ж–µ—Б—Б –∞—В—А–Њ—Д–Є–Є –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞ –њ—А–Є –Я–Ю–£–У –≤—Л—Е–Њ–і–Є—В –Ј–∞ —А–∞–Љ–Ї–Є –≥–ї–∞–Ј–љ–Њ–≥–Њ —П–±–ї–Њ–Ї–∞ [8]. –Я—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є –Ї–Њ–Љ–њ—М—О—В–µ—А–љ–Њ–є —В–Њ–Љ–Њ–≥—А–∞—Д–Є–Є –Њ—А–±–Є—В–∞–ї—М–љ–Њ–є —З–∞—Б—В–Є –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞ —Г –±–Њ–ї—М–љ—Л—Е —Б –Я–Ю–£–У –љ–∞–±–ї—О–і–∞–ї–Њ—Б—М –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ–µ —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –і–Є–∞–Љ–µ—В—А–∞ –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞ (2,4±2,0 –Љ–Љ) –њ–Њ —Б—А–∞–≤–љ–µ–љ–Є—О —Б–Њ –Ј–і–Њ—А–Њ–≤—Л–Љ–Є –ї—О–і—М–Љ–Є (2,8±2,1 –Љ–Љ), —Б–љ–Є–ґ–∞–ї–∞—Б—М –Є –њ–ї–Њ—В–љ–Њ—Б—В—М –Є–Ј–Њ–±—А–∞–ґ–µ–љ–Є—П. Beathy S. —Б —Б–Њ–∞–≤—В. (1998) –њ—А–Њ–≤–Њ–і–Є–ї —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–µ —Б–Ї–∞–љ–Є—А–Њ–≤–∞–љ–Є–µ –Њ—А–±–Є—В–∞–ї—М–љ–Њ–є —З–∞—Б—В–Є –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞ —Г –±–Њ–ї—М–љ—Л—Е –Я–Ю–£–У –Є –≤—Л—П–≤–Є–ї, —З—В–Њ –і–Є–∞–Љ–µ—В—А –µ–≥–Њ —Г–Љ–µ–љ—М—И–∞–ї—Б—П –≤ —Б—А–µ–і–љ–µ–Љ –љ–∞ 0,4 –Љ–Љ –њ—А–Њ—В–Є–≤ –љ–Њ—А–Љ—Л [6].
–Ш–Ј–Љ–µ–љ–µ–љ–Є—П –≤ —Ж–µ–љ—В—А–∞–ї—М–љ–Њ–є —З–∞—Б—В–Є –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞—В–Њ—А–∞ –њ—А–Є –Њ—Д—В–∞–ї—М–Љ–Њ–≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є –±—Л–ї–Є –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–љ—Л –≤ —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–µ —Г –Њ–±–µ–Ј—М—П–љ [15]. –Я—А–Є –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –ґ–Є–≤–Њ—В–љ—Л—Е —Б —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ–Њ–є –≥–ї–∞—Г–Ї–Њ–Љ–Њ–є –≤—Л—П–≤–Є–ї–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ—Г—О –∞—В—А–Њ—Д–Є—О –ї–∞—В–µ—А–∞–ї—М–љ—Л—Е –Ї–Њ–ї–µ–љ—З–∞—В—Л—Е —В–µ–ї (–Ы–Ъ–Ґ), –њ—А–Є—З–µ–Љ –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –∞—В—А–Њ—Д–Є–Є –љ–∞–њ—А—П–Љ—Г—О –Ј–∞–≤–Є—Б–Є—В –Њ—В –і–ї–Є—В–µ–ї—М–љ–Њ—Б—В–Є –Њ—Д—В–∞–ї—М–Љ–Њ–≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є –Є —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ –≤ –і–Є—Б–Ї–µ –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞. –Ф–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П –≤ –Ы–Ъ–Ґ –њ—А–Њ–Є—Б—Е–Њ–і—П—В —Б–љ–∞—З–∞–ї–∞ –≤ –Љ–∞–≥–љ–Њ—Ж–µ–ї–ї—О–ї—П—А–љ–Њ–Љ —Б–ї–Њ–µ, –∞ –Ј–∞—В–µ–Љ –Ј–∞—В—А–∞–≥–Є–≤–∞–µ—В—Б—П –Є –њ–∞—А–≤–Њ—Ж–µ–ї–ї—О–ї—П—А–љ—Л–є —Б–ї–Њ–є. –Ш–Љ–µ–љ–љ–Њ —Н—В–Є–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–Є –Њ–±—К—П—Б–љ—П—О—В –Є–Ј–Љ–µ–љ–µ–љ–Є–µ —Ж–≤–µ—В–Њ–≤–Њ–≥–Њ –Ј—А–µ–љ–Є—П –њ—А–Є –≥–ї–∞—Г–Ї–Њ–Љ–µ. –£–Љ–µ–љ—М—И–µ–љ–Є–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –љ–µ–є—А–Њ–љ–Њ–≤ –±—Л–ї–Њ –Ј–∞—Д–Є–Ї—Б–Є—А–Њ–≤–∞–љ–Њ –≤–њ–ї–Њ—В—М –і–Њ –Ї–Њ—А—Л –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –±—Л–ї–Њ –≤—Л—П–≤–ї–µ–љ–Њ —Б–љ–Є–ґ–µ–љ–Є–µ –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —Н—В–Є—Е –Ї–ї–µ—В–Њ–Ї –њ–Њ —Г—А–Њ–≤–љ—О —Ж–Є—В–Њ—Е—А–Њ–Љ –Њ–Ї—Б–Є–і–∞–Ј—Л [36]. –Р–≤—В–Њ—А—Л –і–Њ–њ—Г—Б–Ї–∞—О—В, —З—В–Њ –њ–Њ—В–µ—А—П –љ–µ—А–≤–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –њ–Њ —Е–Њ–і—Г –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞ –Њ—В –≥–∞–љ–≥–ї–Є–Њ–љ–∞—А–љ—Л—Е –Ї–ї–µ—В–Њ–Ї —Б–µ—В—З–∞—В–Ї–Є –і–Њ –Ї–Њ—А—Л –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Љ–Њ–ґ–µ—В –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞—В—М –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—О –≥–ї–∞—Г–Ї–Њ–Љ—Л, —Б–љ–Є–ґ–∞—В—М –Ј—А–Є—В–µ–ї—М–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є –Є —Г–Љ–µ–љ—М—И–∞—В—М –њ–Њ—А–Њ–≥ –≤–Њ—Б–њ—А–Є–Є–Љ—З–Є–≤–Њ—Б—В–Є –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞ –Ї –њ–Њ–≤—В–Њ—А–љ—Л–Љ –∞–ї—М—В–µ—А–∞—Ж–Є—П–Љ.
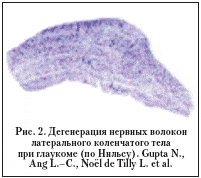
–Ю—Б–Њ–±—Л–є –Є–љ—В–µ—А–µ—Б –≤—Л–Ј—Л–≤–∞—О—В –і–≤–∞ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –љ–∞–±–ї—О–і–µ–љ–Є—П: –Є–Ј—Г—З–∞–ї–Њ—Б—М —Б–Њ—Б—В–Њ—П–љ–Є–µ —Ж–µ–љ—В—А–∞–ї—М–љ–Њ–≥–Њ –Њ—В–і–µ–ї–∞ –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞—В–Њ—А–∞ –±–Њ–ї—М–љ—Л—Е –Я–Ю–£–У –њ–Њ—Б–ї–µ –Є—Е —Б–Љ–µ—А—В–Є, –љ–µ —Б–≤—П–Ј–∞–љ–љ–Њ–є —Б –њ–∞—В–Њ–ї–Њ–≥–Є–µ–є –¶–Э–° (–Њ–і–Є–љ —Б–ї—Г—З–∞–є – –≤ –Ъ–∞–љ–∞–і–µ [20], –і—А—Г–≥–Њ–є – –≤ –°–∞–љ–Ї—В––Я–µ—В–µ—А–±—Г—А–≥–µ [1]). –Я–Њ –њ–Њ–ї—Г—З–µ–љ–љ—Л–Љ –і–∞–љ–љ—Л–Љ, –≤ –Ј—А–Є—В–µ–ї—М–љ–Њ–Љ –љ–µ—А–≤–µ –Љ–∞–Ї—А–Њ—Б–Ї–Њ–њ–Є—З–µ—Б–Ї–Є –љ–∞–±–ї—О–і–∞–ї–∞—Б—М –≤—Л—А–∞–ґ–µ–љ–љ–∞—П –∞—В—А–Њ—Д–Є—П —Б –њ–Њ—В–µ—А–µ–є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –∞–Ї—Б–Њ–љ–Њ–≤. –Ы–Ъ–Ґ –±—Л–ї–Є —Г–Љ–µ–љ—М—И–µ–љ—Л –≤ —А–∞–Ј–Љ–µ—А–∞—Е –≤ —Б–≤—П–Ј–Є —Б –і–µ–≥–µ–љ–µ—А–∞—Ж–Є–µ–є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –љ–µ–є—А–Њ–љ–Њ–≤ (—А–Є—Б. 2). –Я—А–Є –Љ–Є–Ї—А–Њ—Б–Ї–Њ–њ–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ—Л —Г–Љ–µ–љ—М—И–µ–љ–Є–µ —А–∞–і–Є—Г—Б–∞ –љ–µ–є—А–Њ–љ–Њ–≤ –Є –Є—Е —П–і–µ—А, –Ї–Њ–Љ–Ї–Њ–≤–∞—В–∞—П, –Ј–µ—А–љ–Є—Б—В–∞—П —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–∞, –∞ —В–∞–Ї–ґ–µ –±–Њ–ї—М—И–Њ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –њ–Є–≥–Љ–µ–љ—В–∞ –ї–Є–њ–Њ—Д—Г—Б—Ж–Є–љ–∞ – –Њ–і–љ–Њ–≥–Њ –Є–Ј –Љ–∞—А–Ї–µ—А–Њ–≤ –∞—В—А–Њ—Д–Є–Є. –Т –Ј—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ—А–µ –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –±–Њ–ї—М–љ—Л—Е –≤—Л—П–≤–ї–µ–љ–Њ –≤–Є–і–Є–Љ–Њ–µ –і–∞–ґ–µ –љ–µ–≤–Њ–Њ—А—Г–ґ–µ–љ–љ—Л–Љ –≥–ї–∞–Ј–Њ–Љ —Г–Љ–µ–љ—М—И–µ–љ–Є–µ —В–Њ–ї—Й–Є–љ—Л –Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ —Б–ї–Њ—П.
–Э–∞ —Б–Њ–≤—А–µ–Љ–µ–љ–љ–Њ–Љ —Н—В–∞–њ–µ –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–µ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –∞–њ–њ–∞—А–∞—В–Њ–≤ –Љ–∞–≥–љ–Є—В–љ–Њ–—А–µ–Ј–Њ–љ–∞–љ—Б–љ–Њ–є —В–Њ–Љ–Њ–≥—А–∞—Д–Є–Є (–Ь–†–Ґ) –њ–Њ–Ј–≤–Њ–ї—П—О—В –Њ–њ—А–µ–і–µ–ї–Є—В—М —А–∞–Ј–Љ–µ—А—Л –љ–µ —В–Њ–ї—М–Ї–Њ –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞, –љ–Њ –Є –і—А—Г–≥–Є—Е —Б—В—А—Г–Ї—В—Г—А –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞. –Ґ–∞–Ї, –≤ 2009 –≥. F.G. Garaci —Б —Б–Њ–∞–≤—В. [17] –њ–Њ–Ї–∞–Ј–∞–ї –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ–µ —Г–Љ–µ–љ—М—И–µ–љ–Є–µ –і–Є–∞–Љ–µ—В—А–∞ –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞ –Є —Б–љ–Є–ґ–µ–љ–Є–µ –њ–ї–Њ—В–љ–Њ—Б—В–Є —Б–Є–≥–љ–∞–ї–∞ —Б –Њ–±–ї–∞—Б—В–Є –Ј—А–Є—В–µ–ї—М–љ–Њ–є –ї—Г—З–Є—Б—В–Њ—Б—В–Є —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Я–Ю–£–У –љ–∞ —А–∞–Ј–ї–Є—З–љ—Л—Е —Б—В–∞–і–Є—П—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Ч–∞–≤–Є—Б–Є–Љ–Њ—Б—В—М –Є–Ј–Љ–µ–љ–µ–љ–Є–є –Њ—В —Б—В–∞–і–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –±—Л–ї–∞ –Њ—З–µ–љ—М –≤—Л—Б–Њ–Ї–Њ–є, –Ї–Њ—Н—Д—Д–Є—Ж–Є–µ–љ—В –Ї–Њ—А—А–µ–ї—П—Ж–Є–Є –Љ–µ–ґ–і—Г –≥—А—Г–њ–њ–∞–Љ–Є —Б–Њ—Б—В–∞–≤–Є–ї –≤ —Б—А–µ–і–љ–µ–Љ –≥ =0,8087, —А<0,0001. –†–∞–Ј—А–µ—И–µ–љ–Є–µ –Ь–†–Ґ –≤ 3 —В–µ—Б–ї–∞ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Њ –і—А—Г–≥–Є–Љ –∞–≤—В–Њ—А–∞–Љ –љ–µ —В–Њ–ї—М–Ї–Њ –Њ–њ—А–µ–і–µ–ї–Є—В—М –њ–ї–Њ—В–љ–Њ—Б—В—М –Є —А–∞–Ј–Љ–µ—А –Ы–Ъ–Ґ —Г –±–Њ–ї—М–љ—Л—Е —Б –≥–ї–∞—Г–Ї–Њ–Љ–Њ–є, –љ–Њ –Є –Є–Ј–Љ–µ—А–Є—В—М –Є—Е –Њ–±—К–µ–Љ –≤ 2011 –≥. [16]. –°—А–µ–і–љ–Є–µ –Њ–±—К–µ–Љ—Л –Ы–Ъ–Ґ –≤ –Ї–Њ–љ—В—А–Њ–ї—М–љ–Њ–є –≥—А—Г–њ–њ–µ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б–Њ—Б—В–∞–≤–ї—П–ї–Є 98,0±27,2 –Љ–Љ3 (—Б–њ—А–∞–≤–∞) –Є 93,7±25,8 –Љ–Љ3 (—Б–ї–µ–≤–∞), –∞ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –≥–ї–∞—Г–Ї–Њ–Љ–Њ–є –Њ–±—К–µ–Љ –Ы–Ъ–Ґ –±—Л–ї –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –Љ–µ–љ—М—И–µ –Є —Б–Њ—Б—В–∞–≤–ї—П–ї 85,2±27,1 –Є 80,5±23,6 –Љ–Љ3 —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ (—А<0,001). –Ш–Ј–Љ–µ–љ–µ–љ–Є–µ –Њ–±—К–µ–Љ–∞ –Ы–Ъ–Ґ –≤ –≥—А—Г–њ–њ–µ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –≥–ї–∞—Г–Ї–Њ–Љ–Њ–є –Ї–Њ—А—А–µ–ї–Є—А–Њ–≤–∞–ї–Њ —Б–Њ —Б—В–∞–і–Є–µ–є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Р–≤—В–Њ—А—Л –њ—А–µ–і–ї–∞–≥–∞—О—В –і–∞–љ–љ—Л–µ –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–µ –Љ–µ—В–Њ–і–Є–Ї–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –і–ї—П –≤—Л—П–≤–ї–µ–љ–Є—П –≥–ї–∞—Г–Ї–Њ–Љ—Л, —Г—В–Њ—З–љ–µ–љ–Є—П —Б—В–∞–і–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Є –Ї–Њ–љ—В—А–Њ–ї—П –Ј–∞ –і–Є–љ–∞–Љ–Є–Ї–Њ–є –љ–µ–є—А–Њ–њ—А–Њ—В–µ–Ї—В–Є–≤–љ–Њ–є —В–µ—А–∞–њ–Є–Є [23,30].
–У–ї–∞—Г–Ї–Њ–Љ–∞ –Є –±–Њ–ї–µ–Ј–љ—М –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞. –Ю–±—К–µ–Ї—В–Є–≤–љ—Л–Љ –Љ–µ—В–Њ–і–Њ–Љ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞ –љ–∞ —Б–µ–≥–Њ–і–љ—П—И–љ–Є–є –і–µ–љ—М —П–≤–ї—П–µ—В—Б—П –Ь–†–Ґ. –Э–Њ –љ–µ —В–Њ–ї—М–Ї–Њ —Н—В–Њ –і–µ–ї–∞–µ—В –і–∞–љ–љ—Л–µ –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–Њ—Е–Њ–ґ–Є–Љ–Є –і—А—Г–≥ –љ–∞ –і—А—Г–≥–∞. –Ш—Е —Б–±–ї–Є–ґ–∞–µ—В —В–∞–Ї–ґ–µ –≤–Њ–Ј—А–∞—Б—В –њ–∞—Ж–Є–µ–љ—В–Њ–≤, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В–Є –≤ –≤–Њ–Ј—А–∞—Б—В–љ—Л—Е –њ–Њ–њ—Г–ї—П—Ж–Є—П—Е, –Є–Ј–±–Є—А–∞—В–µ–ї—М–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Њ–і–љ–Њ–≥–Њ –≤–Є–і–∞ –љ–µ–є—А–Њ–љ–∞, –Љ–µ—Е–∞–љ–Є–Ј–Љ –≥–Є–±–µ–ї–Є –љ–µ—А–≤–љ–Њ–є –Ї–ї–µ—В–Ї–Є [12,13]. –Т –њ–Њ—Б–ї–µ–і–љ–Є–µ –≥–Њ–і—Л –і–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –Ї–ї—О—З–µ–≤—Г—О —А–Њ–ї—М –≤ –љ–∞—А—Г—И–µ–љ–Є–Є –љ–µ—А–≤–љ–Њ–є –њ–µ—А–µ–і–∞—З–Є –њ—А–Є –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞ –Є–≥—А–∞–µ—В –љ–µ–є—А–Њ—В–Њ–Ї—Б–Є—З–љ—Л–є –±–µ–ї–Њ–Ї β––∞–Љ–Є–ї–Њ–Є–і (–С–Р) [4]. –Я—А–Њ–і—Г–Ї—Ж–Є—П –С–Р –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ—А–Є –Љ—Г—В–∞—Ж–Є–Є –≥–µ–љ–∞, –Ї–Њ–і–Є—А—Г—О—Й–µ–≥–Њ —Б–Є–љ—В–µ–Ј –±–µ–ї–Ї–∞ – –њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞ –∞–Љ–Є–ї–Њ–Є–і–∞, –Є–ї–Є –њ—А–Є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–Љ –і–µ—В–µ—А–Љ–Є–љ–Є—А–Њ–≤–∞–љ–Є–Є. –С–Р –љ–∞—А—Г—И–∞–µ—В –њ–µ—А–µ–і–∞—З—Г –љ–µ—А–≤–љ—Л—Е –Є–Љ–њ—Г–ї—М—Б–Њ–≤ –≤–љ—Г—В—А–Є –Ї–ї–µ—В–Њ–Ї –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞, –≤—Б—В—А–∞–Є–≤–∞—П—Б—М –≤ —Б—В—А—Г–Ї—В—Г—А–љ—Л–µ —Ж–µ–њ–Є —В—А–∞–љ—Б—Д–µ—А–∞ –љ–µ—А–≤–љ–Њ–≥–Њ —Б–Є–≥–љ–∞–ї–∞. –Э–µ–є—А–Њ—В–Њ–Ї—Б–Є—З–љ—Л–є —Н—Д—Д–µ–Ї—В –С–Р —Б–≤—П–Ј–∞–љ —Б –µ–≥–Њ —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М—О –Њ—В–Ї—А—Л–≤–∞—В—М –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ—Л–µ –њ–Њ—А—Л [11]. –Я—А–Є —Н—В–Њ–Љ –љ–∞—А—Г—И–∞–µ—В—Б—П –≥–Њ–Љ–µ–Њ—Б—В–∞–Ј –Ї–∞–ї—М—Ж–Є—П –≤ —Ж–Є—В–Њ–Ј–Њ–ї–µ –љ–µ—А–≤–љ–Њ–є –Ї–ї–µ—В–Ї–Є, –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–Є –љ–∞–±—Г—Е–∞—О—В, –≤—Л—Б–≤–Њ–±–Њ–ґ–і–∞—О—В—Б—П –∞–Ї—В–Є–≤–∞—В–Њ—А—Л –Ї–∞—Б–њ–∞–Ј, –Ј–∞–њ—Г—Б–Ї–∞–µ—В—Б—П –Љ–µ—Е–∞–љ–Є–Ј–Љ –љ–µ–Њ–±—А–∞—В–Є–Љ–Њ–є –≥–Є–±–µ–ї–Є –Ї–ї–µ—В–Ї–Є –њ–Њ –њ—Г—В–Є –∞–њ–Њ–њ—В–Њ–Ј–∞. –Р–њ–Њ–њ—В–Њ–Ј – —Н—В–Њ –Њ—Б–љ–Њ–≤–љ–Њ–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –≥–Є–±–µ–ї–Є –Ї–ї–µ—В–Ї–Є –њ—А–Є –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е.
–£ –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –±–Њ–ї–µ–Ј–љ—М—О –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞ –љ–∞ —Д–Њ–љ–µ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞ –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –Є —Б–љ–Є–ґ–µ–љ–Є–µ –Ј—А–µ–љ–Є—П [24,29]. –Ф–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞—В—М —Н—В–Њ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ —В–Њ–ї—М–Ї–Њ –њ—А–Є –Њ–±—К–µ–Ї—В–Є–≤–љ–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є, —В.–Ї. –і–∞–љ–љ–∞—П –Ї–∞—В–µ–≥–Њ—А–Є—П –±–Њ–ї—М–љ—Л—Е –љ–µ –њ—А–µ–і—К—П–≤–ї—П–µ—В –ґ–∞–ї–Њ–± –љ–∞ –љ–Є–Ј–Ї–Њ–µ –Ј—А–µ–љ–Є–µ. –Ч–∞–±–Њ–ї–µ–≤–∞–µ–Љ–Њ—Б—В—М –Я–Ю–£–У –њ—А–Є –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞ –≤ 5 —А–∞–Ј –≤—Л—И–µ, —З–µ–Љ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —В–Њ–є –ґ–µ –≤–Њ–Ј—А–∞—Б—В–љ–Њ–є –≥—А—Г–њ–њ—Л [14].
–Т –њ–Њ—Б–ї–µ–і–љ–Є–µ –≥–Њ–і—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–Є —Г–і–µ–ї—П—О—В –±–Њ–ї—М—И–Њ–µ –≤–љ–Є–Љ–∞–љ–Є–µ —А–Њ–ї–Є –С–Р –≤ —А–∞–Ј–≤–Є—В–Є–Є –≥–ї–∞—Г–Ї–Њ–Љ–љ–Њ–є –љ–µ–є—А–Њ–Њ–њ—В–Є–Ї–Њ–њ–∞—В–Є–Є [34,35]. –Ґ–∞–Ї, –њ—А–Є –Љ–Њ–і–µ–ї–Є—А–Њ–≤–∞–љ–Є–Є –Њ—Д—В–∞–ї—М–Љ–Њ–≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є —Г –Љ—Л—И–µ–є –±—Л–ї–Њ –≤—Л—П–≤–ї–µ–љ–Њ –њ–Њ–≤—Л—И–µ–љ–љ–Њ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –±–µ–ї–Ї–∞ – –њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞ –С–Р —Г –±–Њ–ї–µ–µ –≤–Ј—А–Њ—Б–ї—Л—Е –Љ—Л—И–µ–є –њ–Њ —Б—А–∞–≤–љ–µ–љ–Є—О —Б –Љ–Њ–ї–Њ–і—Л–Љ–Є –Є –Є–љ—В–∞–Ї—В–љ—Л–Љ–Є –ґ–Є–≤–Њ—В–љ—Л–Љ–Є [18]. –Ф—А—Г–≥–Є–Љ–Є –∞–≤—В–Њ—А–∞–Љ–Є —В–∞–Ї–ґ–µ –±—Л–ї–Њ –≤—Л—П–≤–ї–µ–љ–Њ –њ—А–Є—Б—Г—В—Б—В–≤–Є–µ –С–Р —Г –Љ—Л—И–µ–є —Б —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ–Њ–є –≥–ї–∞—Г–Ї–Њ–Љ–Њ–є, —Г—А–Њ–≤–µ–љ—М –µ–≥–Њ –љ–Њ—Б–Є–ї –і–Њ–Ј–Њ–Ј–∞–≤–Є—Б–Є–Љ—Л–є —Н—Д—Д–µ–Ї—В [26]. –Т–≤–µ–і–µ–љ–Є–µ –≤–µ—Й–µ—Б—В–≤, –±–ї–Њ–Ї–Є—А—Г—О—Й–Є—Е –С–Р (–Є–љ–≥–Є–±–Є—В–Њ—А β–—Б–µ–Ї—А–µ—В–∞–Ј—Л, –∞–љ—В–Є—В–µ–ї–∞ –Ї –∞–љ—В–Є–β––∞–Љ–Є–ї–Њ–Є–і—Г, –∞–љ—В–Є–∞–≥—А–µ–≥–∞–љ—В –С–Р «–Ъ–Њ–љ–≥–Њ –Ї—А–∞—Б–љ—Л–є»), –Њ–Ї–∞–Ј—Л–≤–∞–ї–Њ –љ–µ–є—А–Њ–њ—А–Њ—В–µ–Ї—В–Є–≤–љ—Л–є —Н—Д—Д–µ–Ї—В –њ—А–Є —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ–Њ–є –≥–ї–∞—Г–Ї–Њ–Љ–µ [19]. –Э–∞ –Ї—Г–ї—М—В—Г—А–µ –≤—Л–і–µ–ї–µ–љ–љ—Л—Е –≥–∞–љ–≥–ї–Є–Њ–љ–∞—А–љ—Л—Е –Ї–ї–µ—В–Њ–Ї —Б–µ—В—З–∞—В–Ї–Є –Љ—Л—И–µ–є –±—Л–ї–Њ –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–љ–Њ –љ–µ–є—А–Њ—В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–µ –і–µ–є—Б—В–≤–Є–µ –њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞ –С–Р –≤ –њ—А–Є—Б—Г—В—Б—В–≤–Є–Є –Є–љ–і—Г–Ї—В–Њ—А–∞ —Б–Є–љ—В–µ–Ј–∞ –С–Р. –Ф–µ–≥–µ–љ–µ—А–∞—Ж–Є—П –љ–µ–є—А–Њ–љ–Њ–≤ —Б–µ—В—З–∞—В–Ї–Є –±—Л–ї–∞ –њ—А–Њ–њ–Њ—А—Ж–Є–Њ–љ–∞–ї—М–љ–∞ –≤—А–µ–Љ–µ–љ–Є –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –С–Р –Є –µ–≥–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є. –Я—А–Є –і–Њ–±–∞–≤–ї–µ–љ–Є–Є –≤ –Ї—Г–ї—М—В—Г—А—Г –Ї–ї–µ—В–Њ–Ї –Є–љ–≥–Є–±–Є—В–Њ—А–∞ —Б–Є–љ—В–µ–Ј–∞ –С–Р –≥–Є–±–µ–ї—М –Ї–ї–µ—В–Њ–Ї –њ—А–µ–Ї—А–∞—Й–∞–ї–∞—Б—М [32].
–°—Л–≤–Њ—А–Њ—В–Њ—З–љ—Л–є –∞–Љ–Є–ї–Њ–Є–і –±—Л–ї —В–∞–Ї–ґ–µ –Њ–±–љ–∞—А—Г–ґ–µ–љ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –≥–ї–∞—Г–Ї–Њ–Љ–Њ–є –њ—А–Є –њ–Њ–Љ–Њ—Й–Є –Я–¶–† –≤ —В—А–∞–±–µ–Ї—Г–ї—П—А–љ–Њ–є –Ј–Њ–љ–µ, –∞ —Г—А–Њ–≤–µ–љ—М –µ–≥–Њ –≤ —Б—Л–≤–Њ—А–Њ—В–Ї–µ –±—Л–ї –≤—Л—И–µ, —З–µ–Љ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –±–µ–Ј –≥–ї–∞—Г–Ї–Њ–Љ—Л [33]. –°—Г—Й–µ—Б—В–≤—Г–µ—В –Љ–љ–µ–љ–Є–µ, —З—В–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –≥–ї–∞—Г–Ї–Њ–Љ—Л –љ–∞–њ—А—П–Љ—Г—О —Б–≤—П–Ј–∞–љ–Њ —Б —Г—А–Њ–≤–љ–µ–Љ –С–Р –Є —В–∞—Г––±–µ–ї–Ї–∞ –≤ —Б–њ–Є–љ–љ–Њ–Љ–Њ–Ј–≥–Њ–≤–Њ–є –ґ–Є–і–Ї–Њ—Б—В–Є —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Я–Ю–£–У [27]. –°–Њ–≤—А–µ–Љ–µ–љ–љ–Є–Ї–Є –≤—Б–µ –±–Њ–ї—М—И–µ —Б–Ї–ї–Њ–љ—П—О—В—Б—П –Ї –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –∞–њ—А–Њ–±–∞—Ж–Є–Є –њ—А–µ–њ–∞—А–∞—В–Њ–≤, –њ—А–Є–Љ–µ–љ—П–µ–Љ—Л—Е –≤ –ї–µ—З–µ–љ–Є–Є –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ –њ—А–Є –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞, –≤ —В–µ—А–∞–њ–Є–Є –≥–ї–∞—Г–Ї–Њ–Љ–љ–Њ–є –љ–µ–є—А–Њ–Њ–њ—В–Є–Ї–Њ–њ–∞—В–Є–Є [25].
–Ь–µ—Е–∞–љ–Є–Ј–Љ –≥–Є–±–µ–ї–Є –Ї–ї–µ—В–Ї–Є –њ—А–Є –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е. –С–Њ–ї—М—И–∞—П —З–∞—Б—В—М –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є —П–≤–ї—П–µ—В—Б—П –њ–Њ–ї–Є—Н—В–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є, –Є –≤ –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –≤—Л–і–µ–ї–Є—В—М –њ—Г—Б–Ї–Њ–≤–Њ–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –і–ї—П –Ї–∞–ґ–і–Њ–≥–Њ –і–Њ—Б—В–∞—В–Њ—З–љ–Њ —Б–ї–Њ–ґ–љ–Њ. –Ґ–µ–Љ –љ–µ –Љ–µ–љ–µ–µ —Б—Г—Й–µ—Б—В–≤—Г—О—В —Г–±–µ–і–Є—В–µ–ї—М–љ—Л–µ –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М—Б—В–≤–∞ —В–Њ–≥–Њ, —З—В–Њ —Ж–µ–љ—В—А–∞–ї—М–љ–∞—П —А–Њ–ї—М –≤ –њ—А–Њ—Ж–µ—Б—Б–∞—Е –∞–њ–Њ–њ—В–Њ–Ј–∞ –љ–µ—А–≤–љ–Њ–є –Ї–ї–µ—В–Ї–Є –њ—А–Є–љ–∞–і–ї–µ–ґ–Є—В –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є—П–Љ [10]. –Я—А–Є —А–∞–Ј–ї–Є—З–љ—Л—Е —Б–Њ—Б—В–Њ—П–љ–Є—П—Е (—Б—В–∞—А–µ–љ–Є–µ –Њ—А–≥–∞–љ–Є–Ј–Љ–∞, «–Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л–є —Б—В—А–µ—Б—Б», –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –Љ—Г—В–∞–љ—В–љ–Њ–є –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–Њ–є –Ф–Э–Ъ) –Є –њ—А–Є –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–Є —А–∞–Ј–ї–Є—З–љ—Л—Е –≤–µ—Й–µ—Б—В–≤ (–љ–µ–є—А–Њ—В–Њ–Ї—Б–Є—З–љ—Л—Е –±–µ–ї–Ї–Њ–≤, –≤ —В–Њ–Љ —З–Є—Б–ї–µ –С–Р) –Є–Ј–Љ–µ–љ—П–µ—В—Б—П –њ—А–Њ–љ–Є—Ж–∞–µ–Љ–Њ—Б—В—М –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ—Л—Е –њ–Њ—А [11]. –≠—В–Њ—В –њ—А–Њ—Ж–µ—Б—Б –њ—А–Є–≤–Њ–і–Є—В –Ї –≤—Л–±—А–Њ—Б—Г –Є–Ј –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –Є–Њ–љ–Њ–≤ –Ї–∞–ї—М—Ж–Є—П –Є –∞–Ї—В–Є–≤–∞—В–Њ—А–Њ–≤ –∞–њ–Њ–њ—В–Њ–Ј–∞, —З—В–Њ –Є –Њ–њ—А–µ–і–µ–ї—П–µ—В –љ–µ–Њ–±—А–∞—В–Є–Љ–Њ—Б—В—М –њ—А–Њ—Ж–µ—Б—Б–∞ –≥–Є–±–µ–ї–Є –љ–µ–є—А–Њ—Ж–Є—В–∞.
–°—Г—Й–µ—Б—В–≤—Г–µ—В –Љ–Њ–і–µ–ї—М –і–ї—П —В–µ—Б—В–Є—А–Њ–≤–∞–љ–Є—П –љ–µ–є—А–Њ–њ—А–Њ—В–µ–Ї—В–Є–≤–љ—Л—Е —Б–≤–Њ–є—Б—В–≤ –≤–µ—Й–µ—Б—В–≤, –њ—А–Є–Љ–µ–љ—П–µ–Љ—Л—Е –≤ –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є. –Т –Ї—Г–ї—М—В—Г—А—Г –≤—Л–і–µ–ї–µ–љ–љ—Л—Е –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –і–Њ–±–∞–≤–ї—П—О—В –Љ–Њ—Й–љ—Л–є –љ–µ–є—А–Њ—В–Њ–Ї—Б–Є—З–љ—Л–є –±–µ–ї–Њ–Ї –С–Р [11]. –Ю–љ —П–≤–ї—П–µ—В—Б—П —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–Љ –Є —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–Љ –Є–љ–і—Г–Ї—В–Њ—А–Њ–Љ –Њ—В–Ї—А—Л—В–Є—П –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ—Л—Е –њ–Њ—А. –Ю—В–Ї—А—Л—В–Є–µ –њ–Њ—А –Є–Ј—Г—З–∞—О—В –њ–Њ –љ–∞–±—Г—Е–∞–љ–Є—О –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –љ–∞ —Б–њ–µ–Ї—В—А–Њ–Љ–µ—В—А–µ. –Х—Б—В–µ—Б—В–≤–µ–љ–љ—Л–Љ —Н–љ–і–Њ–≥–µ–љ–љ—Л–Љ –љ–µ–є—А–Њ–њ—А–Њ—В–µ–Ї—В–Њ—А–Њ–Љ —П–≤–ї—П–µ—В—Б—П —Е—А–Њ–љ–Њ–±–Є–Њ—В–Є–Ї –Љ–µ–ї–∞—В–Њ–љ–Є–љ. –Х–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ —Г–Љ–µ–љ—М—И–∞–µ—В—Б—П —Б –≤–Њ–Ј—А–∞—Б—В–Њ–Љ, –≤–µ—А–Њ—П—В–љ–Њ, –њ—А–Њ–≤–Њ—Ж–Є—А—Г—П –≤–Њ–Ј—А–∞—Б—В–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Њ—А–≥–∞–љ–Є–Ј–Љ–∞. –Т –Њ–њ—Л—В–∞—Е –љ–∞ –≤—Л–і–µ–ї–µ–љ–љ—Л—Е –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є—П—Е –≤ –њ—А–Є—Б—Г—В—Б—В–≤–Є–Є –њ–∞—А–Ї–Є–љ—Б–Њ–љ–Њ–≥–µ–љ–љ–Њ–≥–Њ –љ–µ–є—А–Њ—В–Њ–Ї—Б–Є–љ–∞ –Љ–µ–ї–∞—В–Њ–љ–Є–љ –њ—А–µ–Ї—А–∞—Й–∞–ї –Є–љ–і—Г–Ї—Ж–Є—О –Њ—В–Ї—А—Л—В–Є—П –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ—Л—Е –њ–Њ—А –Є –љ–∞–±—Г—Е–∞–љ–Є–µ –і–∞–љ–љ—Л—Е –Њ—А–≥–∞–љ–µ–ї–ї.
–Т 2003 –≥. –њ–Њ–і —А—Г–Ї–Њ–≤–Њ–і—Б—В–≤–Њ–Љ –∞–Ї–∞–і–µ–Љ–Є–Ї–∞ –Т.–Я. –°–Ї—Г–ї–∞—З–µ–≤–∞ –љ–∞—З–∞–ї–∞—Б—М —А–∞–Ј—А–∞–±–Њ—В–Ї–∞ –љ–Њ–≤–Њ–≥–Њ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–Њ––∞–і—А–µ—Б–Њ–≤–∞–љ–љ–Њ–≥–Њ –∞–љ—В–Є–Њ–Ї—Б–Є–і–∞–љ—В–∞. –°—А–µ–і–Є —В–µ—Б—В–Є—А—Г–µ–Љ—Л—Е —Б–Њ–µ–і–Є–љ–µ–љ–Є–є – –ї–Є–њ–Њ—Д–Є–ї—М–љ—Л–µ –Ї–∞—В–Є–Њ–љ—Л (–љ–∞–њ—А–Є–Љ–µ—А, –Є–Њ–љ—Л —Д–Њ—Б—Д–Њ–љ–Є—П), —Б–њ–Њ—Б–Њ–±–љ—Л–µ –∞–і—А–µ—Б–љ–Њ –њ—А–Њ–љ–Є–Ї–∞—В—М –≤ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–Є, –і–≤–Є–ґ–Є–Љ—Л–µ —Н–ї–µ–Ї—В—А–Є—З–µ—Б–Ї–Є–Љ –њ–Њ–ї–µ–Љ –љ–∞ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–Њ–є –Љ–µ–Љ–±—А–∞–љ–µ. –≠—В–Є —А–∞–Ј—А–∞–±–Њ—В–Ї–Є –Њ—Б–љ–Њ–≤–∞–љ—Л –љ–∞ —Е–µ–Љ–Є–Њ—Б–Љ–Њ—В–Є—З–µ—Б–Ї–Њ–є –≥–Є–њ–Њ—В–µ–Ј–µ –Я. –Ь–Є—В—З–µ–ї–ї–∞, –њ–Њ—Б—В—Г–ї–Є—А–Њ–≤–∞–≤—И–µ–≥–Њ –љ–∞–ї–Є—З–Є–µ —А–∞–Ј–љ–Њ—Б—В–Є —Н–ї–µ–Ї—В—А–Є—З–µ—Б–Ї–Є—Е –њ–Њ—В–µ–љ—Ж–Є–∞–ї–Њ–≤ –љ–∞ –Љ–µ–Љ–±—А–∞–љ–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є. –Т 1974 –≥. —В–∞–Ї–Є–µ —Б–Њ–µ–і–Є–љ–µ–љ–Є—П –±—Л–ї–Є –љ–∞–Ј–≤–∞–љ—Л –Є–Ј–≤–µ—Б—В–љ—Л–Љ –∞–Љ–µ—А–Є–Ї–∞–љ—Б–Ї–Є–Љ –±–Є–Њ—Е–Є–Љ–Є–Ї–Њ–Љ –Ф. –У—А–Є–љ–Њ–Љ «–Є–Њ–љ–∞–Љ–Є –°–Ї—Г–ї–∞—З–µ–≤–∞». –С—Л–ї–Њ —Б–Ї–Њ–љ—Б—В—А—Г–Є—А–Њ–≤–∞–љ–Њ –Є —Б–Є–љ—В–µ–Ј–Є—А–Њ–≤–∞–љ–Њ –≤–µ—Й–µ—Б—В–≤–Њ SkQ1, —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –Ї–Њ—В–Њ—А–Њ–≥–Њ –Њ–Ї–∞–Ј–∞–ї–∞—Б—М –≤—Л—И–µ –њ—А–µ–і—Л–і—Г—Й–Є—Е –∞–љ–∞–ї–Њ–≥–Њ–≤ –≤ —Б–Њ—В–љ–Є —А–∞–Ј [3].
–Т—Л—Б–Њ–Ї–∞—П —В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –≥–ї–∞–Ј–љ—Л—Е –Ї–∞–њ–µ–ї—М, —Б–Њ–і–µ—А–ґ–∞—Й–Є—Е –Љ–Є—В–Њ—Е–Њ–і—А–Є–∞–ї—М–љ–Њ––∞–і—А–µ—Б–Њ–≤–∞–љ–љ—Л–є –∞–љ—В–Є–Њ–Ї—Б–Є–і–∞–љ—В, –њ–Њ–Ї–∞–Ј–∞–љ–∞ –њ—А–Є —Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ–Њ–є –≥–ї–∞—Г–Ї–Њ–Љ–µ. –£ –Ї—А–Њ–ї–Є–Ї–Њ–≤ —Б –Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є –≥–ї–∞—Г–Ї–Њ–Љ–Њ–є –њ–Њ—Б–ї–µ –Є–љ—Б—В–Є–ї–ї—П—Ж–Є–Є –њ–ї–∞—Б—В–Њ—Е–Є–љ–Њ–љ–Є–ї–і–µ—Ж–Є–ї—В—А–Є—Д–µ–љ–Є–ї—Д–Њ—Б—Д–Њ–љ–Є—П –±—А–Њ–Љ–Є–і–∞ (–Я–Ф–Ґ–§) –Т–У–Ф —Б–љ–Є–ґ–∞–ї–Њ—Б—М –њ–Њ —Б—А–∞–≤–љ–µ–љ–Є—О —Б –Є–љ—В–∞–Ї—В–љ—Л–Љ–Є –≥–ї–∞–Ј–∞–Љ–Є. –Ф–∞–љ–љ–Њ–µ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–Њ––љ–∞–њ—А–∞–≤–ї–µ–љ–љ–Њ–µ —Б–Њ–µ–і–Є–љ–µ–љ–Є–µ –Њ–Ї–∞–Ј—Л–≤–∞–ї–Њ –Є –≤—Л—А–∞–ґ–µ–љ–љ—Л–є –љ–µ–є—А–Њ–њ—А–Њ—В–µ–Ї—В–Є–≤–љ—Л–є —Н—Д—Д–µ–Ї—В –љ–∞ –∞–Ї—Б–Њ–љ—Л –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞. –Ю—В–Љ–µ—З–µ–љ–∞ –њ–Њ–ї–љ–∞—П —Б–Њ—Е—А–∞–љ–љ–Њ—Б—В—М –∞–Ї—Б–Њ–љ–Њ–≤ –њ—А–µ–ї–∞–Љ–Є–љ–∞—А–љ–Њ–є –Ј–Њ–љ—Л –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞. –Я–Ф–Ґ–§ –ї–µ–≥–Ї–Њ –њ—А–Њ–љ–Є–Ї–∞–µ—В —З–µ—А–µ–Ј –±–Є—Б–ї–Њ–є–љ—Г—О —Д–Њ—Б—Д–Њ–ї–Є–њ–Є–і–љ—Г—О –Љ–µ–Љ–±—А–∞–љ—Г –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є, —Н–ї–µ–Ї—В—А–Њ—Д–Њ—А–µ—В–Є—З–µ—Б–Ї–Є –љ–∞–Ї–∞–њ–ї–Є–≤–∞–µ—В—Б—П –љ–∞ –≤–љ—Г—В—А–µ–љ–љ–µ–є –Љ–µ–Љ–±—А–∞–љ–µ –Є –Њ—В–ї–Є—З–∞–µ—В—Б—П –≤—Л—Б–Њ–Ї–Њ–є –∞–љ—В–Є–Њ–Ї—Б–Є–і–∞–љ—В–љ–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О [9].
–Т–Њ–Ј–Љ–Њ–ґ–љ–Њ, –≤ —Б–Ї–Њ—А–Њ–Љ –±—Г–і—Г—Й–µ–Љ –Љ–Є—И–µ–љ—М—О –і–ї—П –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –≤—Б–µ—Е –љ–µ–є—А–Њ–њ—А–Њ—В–µ–Ї—В–Є–≤–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ —Б—В–∞–љ—Г—В –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–Є. –Ъ–∞–Ї –Є –≤ –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є, –≤ –Њ—Д—В–∞–ї—М–Љ–Њ–ї–Њ–≥–Є–Є –љ–µ –њ—А–µ–Ї—А–∞—Й–∞–µ—В—Б—П –њ–Њ–Є—Б–Ї –∞–і–µ–Ї–≤–∞—В–љ—Л—Е —Б—А–µ–і—Б—В–≤ –і–ї—П –њ—А–µ–і–Њ—В–≤—А–∞—Й–µ–љ–Є—П —Н–≤–Њ–ї—О—Ж–Є–Є –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є.
–Т—Л–≤–Њ–і. –Т—Б–µ –±–Њ–ї—М—И–µ –ї–Є—В–µ—А–∞—В—Г—А–љ—Л—Е –і–∞–љ–љ—Л—Е —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ —В–Њ–Љ, —З—В–Њ –њ—А–Є –Я–Ю–£–У –≤ –њ—А–Њ—Ж–µ—Б—Б –≤–Њ–≤–ї–µ—З–µ–љ—Л –љ–µ —В–Њ–ї—М–Ї–Њ –≥–∞–љ–≥–ї–Є–Њ–љ–∞—А–љ—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞ —Б–µ—В—З–∞—В–Ї–Є –Є –і–Є—Б–Ї –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞, –љ–Њ –Є —Ж–µ–љ—В—А–∞–ї—М–љ—Л–µ –Њ—В–і–µ–ї—Л –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞—В–Њ—А–∞. –Т—Б–µ —Н—В–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –љ–∞–њ—А–∞–≤–ї–µ–љ—Л –љ–∞ –Є–Ј—Г—З–µ–љ–Є–µ –њ—А–Є—З–Є–љ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –≥–ї–∞—Г–Ї–Њ–Љ–љ–Њ–є –љ–µ–є—А–Њ–Њ–њ—В–Є–Ї–Њ–њ–∞—В–Є–Є –Є —Б–љ–Є–ґ–µ–љ–Є—П –Ј—А–µ–љ–Є—П. –Т–Њ–Ј–Љ–Њ–ґ–љ–Њ, —Б–Њ–≤–Љ–µ—Б—В–љ–∞—П —А–∞–±–Њ—В–∞ —Б —Г—З–µ–љ—Л–Љ–Є, –Ј–∞–љ–Є–Љ–∞—О—Й–Є–Љ–Є—Б—П –њ—А–Њ–±–ї–µ–Љ–∞–Љ–Є —А–∞–Ј–≤–Є—В–Є—П –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞, –њ–Њ–Љ–Њ–ґ–µ—В –љ–∞–є—В–Є —Б–њ–Њ—Б–Њ–±—Л –њ—А–µ–і–Њ—В–≤—А–∞—В–Є—В—М —Н–≤–Њ–ї—О—Ж–Є—О –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –њ—А–Є –≥–ї–∞—Г–Ї–Њ–Љ–µ.
–Ы–Є—В–µ—А–∞—В—Г—А–∞
1. –Р–ї–µ–Ї—Б–µ–µ–≤ –Т.–Э., –У–∞–Ј–Є–Ј–Њ–≤–∞ –Ш.–†. –Э–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П —Г –±–Њ–ї—М–љ—Л—Е –њ–µ—А–≤–Є—З–љ–Њ–є –Њ—В–Ї—А—Л—В–Њ—Г–≥–Њ–ї—М–љ–Њ–є –≥–ї–∞—Г–Ї–Њ–Љ–Њ–є // –Я—А–∞–Ї—В–Є—З–µ—Б–Ї–∞—П –Љ–µ–і–Є—Ж–Є–љ–∞. 2012.
2. –Р–ї–µ–Ї—Б–µ–µ–≤ –Т.–Э., –У–∞–Ј–Є–Ј–Њ–≤–∞ –Ш.–†., –Э–Є–Ї–Є—В–Є–љ –Ф.–Э –Э–∞—А—Г—И–µ–љ–Є–µ —Н–љ–µ—А–≥–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –Њ–±–Љ–µ–љ–∞ –Ї–ї–µ—В–Ї–Є –Ї–∞–Ї —Д–∞–Ї—В–Њ—А –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –≥–ї–∞—Г–Ї–Њ–Љ–љ–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞ // –Я—А–Њ—Д–Є–ї–∞–Ї—В–Є—З–µ—Б–Ї–∞—П –Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Љ–µ–і–Є—Ж–Є–љ–∞. –Ь–∞—В–µ—А–Є–∞–ї—Л –•IV –†–Њ—Б. –љ–∞—Ж. –Ї–Њ–љ–≥—А–µ—Б—Б–∞ «–І–µ–ї–Њ–≤–µ–Ї –Є –µ–≥–Њ –Ј–і–Њ—А–Њ–≤—М–µ». –°–Я–±., 2011. –°. 12–15.
3. –Р—А—Е–Є–њ–Њ–≤–∞ –Ы.–Ґ., –Р—А—Е–Є–њ–Њ–≤–∞ –Ь.–Ь., –С–∞–Ї–µ–µ–≤–∞ –Ы.–Х. –Є –і—А. –Я—А–Њ–Є–Ј–≤–Њ–і–љ–Њ–µ –њ–ї–∞—Б—В–Њ—Е–Є–љ–Њ–љ–∞, –∞–і—А–µ—Б–Њ–≤–∞–љ–љ–Њ–µ –≤ –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–Є, –Ї–∞–Ї —Б—А–µ–і—Б—В–≤–Њ, –њ—А–µ—А—Л–≤–∞—О—Й–µ–µ –њ—А–Њ–≥—А–∞–Љ–Љ—Г —Б—В–∞—А–µ–љ–Є—П. –°–≤—П–Ј–∞–љ–љ—Л–µ —Б –≤–Њ–Ј—А–∞—Б—В–Њ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –≥–ї–∞–Ј. SkQ –≤–Њ–Ј–≤—А–∞—Й–∞–µ—В –Ј—А–µ–љ–Є–µ —Б–ї–µ–њ—Л–Љ –ґ–Є–≤–Њ—В–љ—Л–Љ // –С–Є–Њ—Е–Є–Љ–Є—П. 2008. –Ґ. 73, вДЦ 12. –°. 1641–1654.
4. –С–∞—З—Г—А–Є–љ –°.–Ю., –®–µ–≤—Ж–Њ–≤–∞ –Х.–§. –Т–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Љ–Є—И–µ–љ–Є –і–ї—П —Б–Њ–Ј–і–∞–љ–Є—П –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –і–ї—П –ї–µ—З–µ–љ–Є—П –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞: –љ–Њ–≤—Л–µ –∞—Б–њ–µ–Ї—В—Л // –Я—Б–Є—Е–Є–∞—В—А–Є—П. 2008. вДЦ 4–6. –°. 43–47.
5. –С–Є–љ–≥ –†., –С—А—Г–Ї–љ–µ—А –†. –Ь–Њ–Ј–≥ –Є –≥–ї–∞–Ј. –Ю—Б–љ–Њ–≤—Л –Њ—Д—В–∞–ї—М–Љ–Њ–љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є / –Я–Њ–і —А–µ–і. –Х.–Ц. –Ґ—А–Њ–љ–∞. –Ы.: –Ь–µ–і–≥–Є–Ј, 1959. –°. 291.
6. –Т–Њ–ї–Ї–Њ–≤ –Т.–Т. –У–ї–∞—Г–Ї–Њ–Љ–∞ –њ—А–Є –њ—Б–µ–≤–і–Њ–љ–Њ—А–Љ–∞–ї—М–љ–Њ–Љ –і–∞–≤–ї–µ–љ–Є–Є: –†—Г–Ї–Њ–≤–Њ–і—Б—В–≤–Њ –і–ї—П –≤—А–∞—З–µ–є. –Ь.: –Ь–µ–і–Є—Ж–Є–љ–∞, 2001. 352 —Б.
7. –У–∞–Ј–Є–Ј–Њ–≤–∞ –Ш.–†. –Ь–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–∞—П –њ–∞—В–Њ–ї–Њ–≥–Є—П –Є –≥–ї–∞—Г–Ї–Њ–Љ–∞ // –У–ї–∞—Г–Ї–Њ–Љ–∞. 2011. вДЦ 4. –°. 58–65.
8. –Ф–Њ–ї–ґ–Є—З –У.–Ш., –£—И–љ–Є–Ї–Њ–≤ –Р.–Э., –Р–±—Г –•–∞–є—А –Э.–Р., –Ю—А–µ—Е–Њ–≤ –Х.–†. –Ъ–Њ–Љ–њ—М—О—В–µ—А–љ–∞—П —В–Њ–Љ–Њ–≥—А–∞—Д–Є—П –Ј—А–Є—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ—А–≤–∞ –њ—А–Є –≥–ї–∞—Г–Ї–Њ–Љ–µ // –°–±. –љ–∞—Г—З. —В—А—Г–і–Њ–≤ «–У–ї–∞—Г–Ї–Њ–Љ–∞». –Т—Л–њ. 2. –Ь., 1996. –°. 107–111.
9. –Ш–Њ–Љ–і–Є–љ–∞ –Х.–Э., –°–µ–љ–Є–љ –Ш.–Ш., –•–Њ—А–Њ—И–Є–ї–Њ–≤–∞––Ь–∞—Б–ї–Њ–≤–∞ –Ш.–Я. –Є –і—А. –Ф–Њ–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –±–µ–Ј–Њ–њ–∞—Б–љ–Њ—Б—В–Є –Є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–Њ––љ–∞–њ—А–∞–≤–ї–µ–љ–љ–Њ–≥–Њ –∞–љ—В–Є–Њ–Ї—Б–Є–і–∞–љ—В–∞ –і–ї—П –њ—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–Є –Є –ї–µ—З–µ–љ–Є—П –≥–ї–∞—Г–Ї–Њ–Љ—Л // –°–±. —Б—В–∞—В–µ–є IX –Љ–µ–ґ–і—Г–љ–∞—А. –Ї–Њ–љ—Д. «–У–ї–∞—Г–Ї–Њ–Љ–∞: —В–µ–Њ—А–Є—П, —В–µ–љ–і–µ–љ—Ж–Є–Є, —В–µ—Е–љ–Њ–ї–Њ–≥–Є–Є». –Ь., 2011. –°. 111–119.
10.–°—Г–і–∞–Ї–Њ–≤ –Э.–Я., –Э–Є–Ї–Є—Д–Њ—А–Њ–≤ –°.–С., –Ъ–Њ–љ—Б—В–∞–љ—В–Є–љ–Њ–≤ –Ѓ.–Ь., –Ы–µ–њ–µ—Е–Њ–≤–∞ –°.–Р. –†–Њ–ї—М –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–є –≤ —А–µ–∞–ї–Є–Ј–∞—Ж–Є–Є –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ –њ—А–Њ–≥—А–∞–Љ–Љ–Є—А–Њ–≤–∞–љ–љ–Њ–є –≥–Є–±–µ–ї–Є –Ї–ї–µ—В–Ї–Є // –С—О–ї–ї–µ—В–µ–љ—М –Т–°–Э–¶ –°–Ю –†–Р–Ь–Э. 2007. вДЦ 1 (53). –°. 103–107.
11. –®–µ–≤—Ж–Њ–≤–∞ –Х.–§., –Ъ–Є—А–µ–µ–≤–∞ –Х.–У., –С–∞—З—Г—А–Є–љ –°.–Ю. –Ь–Є—В–Њ—Е–Њ–љ–і—А–Є–Є –Ї–∞–Ї –Љ–Є—И–µ–љ—М –і–µ–є—Б—В–≤–Є—П –љ–µ–є—А–Њ–њ—А–Њ—В–µ–Ї—В–Њ—А–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤ // –Т–µ—Б—В. –†–Њ—Б. –Р–Ь–Э. 2005. вДЦ 9. –°. 13–17.
12. Bayer A.U., Keller O.N., Ferrari F., Maag K.P. Association of glaucoma with neurodegenerative diseases with apoptotic cell death: Alzheimer's disease and Parkinson's disease // Am. J. Ophthalmol. 2002. вДЦ 1. –†. 135–137.
13. Bizrah M., Guo L., Cordeiro M.F. Glaucoma and Alzheimer's disease in the elderly //Aging Health. 2011. вДЦ 5. –†. 719–733.
14. Blanks J.C., Schmidt S.Y., Torigoe Y. et al. Retinal pathology in Alzheimer's disease. II. Regional neuron loss and glial changes in GCL // Neurobiol. Aging. 1996. вДЦ 3. –†. 385–395.
15. Crawford M.L., Harwerth R.S., Smith E.L. et al. Glaucoma in primates: cytochrome oxidase reactivity in parvo– and magnocellular pathways // Invest. Ophthal. Vis. Sci. 2000. вДЦ 41. –†. 1791–1802.
16. Dai H., Mu K.T., Qi J.P. et al. Assessment of Lateral Geniculate Nucleus Atrophy with 3T MR Imaging and Correlation with Clinical Stage of Glaucoma // Am. J. Neuroradiol. 2011. вДЦ 32. –†. 1347–1353.
17. Garaci F.G., Bolacchi F., Cerulli A. et al. Optic Nerve and Optic Radiation Neurodegeneration in Patients with Glaucoma: In Vivo Analysis with 3–T Diffusion–Tensor MR Imaging // Radiol. 2009. вДЦ 2. –†. 496–501.
18. Goldblum D., Kipfer–Kauer A., Sarra G.M. et al. Distribution of amyloid precursor protein and amyloid–beta immunoreactivity in DBA/2J glaucomatous mouse retinas // Inves. Ophthalmol. & Vis. Sci. 2007. вДЦ 11. –†. 5085–5090.
19. Guo L., Salt Th.E., Luong V., Wood N. Targeting amyloid–b in glaucoma treatment // PNAS. 2007. вДЦ 14. –†. 13444–13449.
20. Gupta N., Ang L–C., Noel de Tilly L. et al. Human glaucoma and neural degeneration in intracranial optic nerve, lateral geniculate nucleus, and visual cortex // Br. J. Ophthalmol. 2006. вДЦ 90. –†. 674–678.
21. Gupta N., Yucel Y.H. Glaucoma and the brain // J. Glaucoma. 2001. вДЦ 10. –†. 28–29.
22. Gupta N., Yucel Y.H. Glaucoma as a neurodegenerative disease // Curr. Opin. Ophthalmol. 2007. вДЦ 2. –†. 110–114.
23. Iba–Zizen M.T., Istoc A., Cabanis E.A. The results of MRI exploration of glaucoma patients: what are the benefits? // Fr. Ophtalmol. 2008. вДЦ 6. –†. 24–28.
24. Iseri P.K., Altinas O., Tokay T., Yuksel N. Relationship between Cognitive Impairment and Retinal Morphological and Visual Functional Abnormalities in Alzheimer Disease // J. Neuroophthalmol. 2006. вДЦ 26 (1). –†. 18–24.
25. Jiahua F., Fagang J., Jingbo L. et al. Rationale for the use of multifunctional drugs as neuroprotective agents for glaucoma // Neural Regenerat. Res. 2012. вДЦ 4. –†. 313–318.
26. McKinnon S.J., Lehman D.M., Kerrigan–Baumrind L.A. et al. Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension // Invest. Ophthalmol. Visual. Sci. 2002. вДЦ 43. –†. 1077–1087.
27. Nucci C., Martucci A., Martorana A. et al. Glaucoma progression associated with altered cerebral spinal fluid levels of amyloid beta and tau proteins // Clin. & Experim. Ophthalmol. 2011. вДЦ3. –†. 279–281.
28. Oliver J.E., Hattenhauer M.G., Herman D. et al. Blindness and glaucoma: a comparison of patients progressing to blindness from glaucoma with patients maintaining vision // Am. J. Ophthalmol. 2002. вДЦ 133. –†. 764–772.
29. Parisi V., Restuccia R., Fattapposta F. et al. Morphological and functional retinal impairment in Alzheimer's disease patients // Clin. Neurophysiol. 2001. вДЦ 112. –†. 1860–1867.
30. Schneider K.A., Richter M.C., Kastner S. Retinotopic organization and functional subdivisions of the human lateral geniculate nucleus: a high–resolution functional magnetic resonance imaging study // J. Neurosci. 2004. вДЦ 24. –†. 8975–8985.
31. Tatton W.G., Chalmers–Redman R.M., Tatton N.A. Apoptosis and anti–apoptosis signalling in glaucomatous retinopathy // Eur. J. Ophthalmol. 2001. Vol. 11, N. 2. P. 12–22.
32. Tsuruma K., Tanaka Y., Shimazawa M., Hara H. Induction of amyloid precursor protein by the neurotoxic peptide, amyloid–beta 25–35, causes retinal ganglion cell death // J. of Neurochem. 2010. вДЦ6. –†. 1545–1554.
33. Wang W.H., McNatt L.G., Pang I.H. et al. Increased expression of serum amyloid A in glaucoma and its effect on intraocular pressure // Inves. Ophthalmol. & Vis. Sci. 2008. вДЦ5. –†. 1916–1923.
34. Yin H., Chen L., Chen X., Liu X. Soluble amyloid beta oligomers may contribute to apoptosis of retinal ganglion cells in glaucoma // Med. Hypoth. 2008. вДЦ 1. –†. 77–80.
35. Yoneda S., Hara H., Hirata A. et al. Vitreous fluid levels of beta–amyloid(1–42) and tau in patients with retinal diseases. // Jpn. J. Ophthalmol. 2005. вДЦ 2. –†. 106–108.
36. Yucel Y.H., Zhang Q., Weinreb R.N. et al. Atrophy of relay neurons in magno– and parvocellular layers in the lateral geniculate nucleus in experimental glaucoma // Invest. Ophthalmol. Vis. Sci. 2001. вДЦ 42. –†. 3216–3222.



