




Ключевые слова: нейродегенерация, глаукомная оптическая нейропатия, бета-амилоид, тау-белок.
Abstract
New molecular and biological aspects
of neurodegeneration in glaucoma. Literature review
Alekseev V.N., Gazizova I.R.
Northwestern State Medical University named after I.I. Mechnikov, St.-Petersburg
The article highlights the issues of neurodegenerative changes in primary open-angle glaucoma. Basing on literature and our own clinical and experimental data we define glaucoma as a neurodegenerative disease that affects not only the retinal ganglion fibers and axons of the optic nerve, but also the pathways of the visual analyzer up to the cerebral cortex. The questions of the impact of neurotoxic protein beta-amyloid and tau-protein on the development and progression of glaucomatous optic neuropathy are discussed.
Key words: neurodegeneration, glaucomatous optic neuropathy, beta-amyloid, tau protein.
Первичная открытоугольная глаукома (ПОУГ) – заболевание, которое развивается с возрастом и характеризуется прогрессивным течением даже на фоне нормализованного уровня офтальмотонуса [1, 3, 8]. Как известно, механизм гибели клеток сетчатки и аксонов зрительного нерва при глаукоме, как и при всех нейродегенеративных расстройствах, – это физиологически запрограммированный апоптоз [2, 9, 54]. Нейродегенерация любого органа характеризуется повреждением клеток и межклеточного вещества, в результате чего изменяется функция органа. В основе ее лежит нарушение трофики, т. е. комплекса механизмов, обеспечивающих метаболизм и сохранность структуры клеток и тканей. Нейродегенеративные заболевания – это заболевания, возникающие в результате прогрессирующей дегенерации и гибели нейронов, входящих в определенные структуры ЦНС, приводящие к разрыву связей между отделами ЦНС и дисбалансу в синтезе и выделении соответствующих нейромедиаторов и, как следствие, вызывающие нарушение памяти, координации движений и мыслительных способностей человека и т. д.
Помимо указанных признаков нейродегенеративные заболевания обладают рядом общих свойств. В частности, подавляющее большинство нейродегенеративных заболеваний развивается у лиц пожилого возраста. Так, например, у пациентов в возрасте 70–75 лет распространенность этих заболеваний составляет около 5%, а в возрасте старше 80 лет достигает 15%. Данные современных клинических и экспериментальных исследований показывают, что основу большинства нейродегенеративных заболеваний определяют генетические факторы (т. е. болезнь или передается по наследству, или возникает в результате патологической прижизненной мутации соответствующих генов). К числу общих признаков нейродегенеративных заболеваний нужно отнести длительный латентный период в их развитии (от 6 до 8–10 лет).
Наиболее известными представителями этого класса заболеваний человека являются болезни Альцгеймера, Паркинсона, Гентингтона, Пика. Поскольку в развитых странах мира наблюдается неуклонное старение населения, общая частота нейродегенеративных заболеваний имеет четкую тенденцию к увеличению [7]. Выделяют заболевания, преимущественно проявляющиеся деменцией: например, болезнь Альцгеймера (атрофируются серое вещество головного мозга и холинэргические нейроны, а страдают в первую очередь когнитивные функции), болезнь Пика – злокачественное слабоумие, при которой происходит атрофия лобных и височных частей коры. Также выделяют заболевания с экстрапирамидными синдромами, например, болезнь Паркинсона, при этом происходит нейродегенерация черной субстанции и дофаминовых нейронов, которая проявляется в виде двигательных нарушений и тремора. При болезни Гентингтона атрофии подвергаются стриатум и кора, что проявляется гиперкинезом и слабоумием. Отдельно выделяют дегенерации с поражением двигательных нейронов, например, боковой амиотрофический склероз, который возникает вследствие дегенерации моторной коры и проявляется в виде паралича и атрофии мышц.
В последнее время доказано, что при глаукоме нейродегенеративным изменениям также подвергаются все зрительные нейроны и зрительная кора головного мозга (рис. 1). Открытым остается вопрос о механизмах нейродегенерации при глаукомной оптической нейропатии [3, 4, 6]. Теория А.П. Нестерова о влиянии аксоплазматического тока на нейродегенерацию аксонов зрительного нерва подтверждается тем, что изменения при глаукоме выходят далеко за рамки глазного яблока. В литературе все чаще встречаются работы, свидетельствующие о наличии тесных связей первичной глаукомы с такими нейродегенеративными заболеваниями, как болезнь Альцгеймера и болезнь Паркинсона [4, 13, 32, 53]. Несомненно, между ними есть много общего: рост численности заболеваемости с возрастом, избранное поражение определенного вида нейронов, один и тот же механизм гибели нервной клетки. Патогенез болезни Альцгеймера до конца не изучен, как и в случае с ПОУГ, но значимым фактором прогрессирования нейродегенерации при данном заболевании считается наличие измененных протеинов: бета-амилоида и тау-белка [45].
В данном обзоре литературы обобщены данные о влиянии бета-амилоида и тау-белка на развитие нейродегенеративного процесса при ПОУГ.
О значении бета-амилоида. Как известно, бета-амилоид является важным звеном патогенеза болезни Альцгеймера. Этот белок образуется при ферментном расщеплении предшественника бета-амилоида APP (Amyloid precursor protein), который является трансмембранным белком. В процессе протеолиза АРР происходят скопление и агрегация нейротоксичной формы бета-амилоида. Отложение фибрилл и аморфных конгломератов бета-амилоида приводит к образованию внеклеточных амилоидных бляшек. Скопление бета-амилоида способствует нарушению межсинаптической нейрональной передачи и формированию вторичной трансинаптической нейродегенерации, разрастанию астроцитов и активации нейроглии. В организме человека ген APP расположен на хромосоме 21 в виде трех основных изоформ (АРР695, АРР751 и АРР770) [25]. Экспрессия APP происходит в высших отделах головного мозга и затем транспортируется антеградно в нейроны (например, от зрительной коры до ганглионарных клеток сетчатки) [14]. Биологическая функция АРР заключается в модуляции роста нейронов [36], синаптической активности [33, 56, 63] и межсинаптической передачи [72]. В сетчатке он необходим для полноценного ретиногенеза [21]. В эксперименте на мышах было показано, что выключение экспрессии АРР приводит к нарушению дифференциации клеток сетчатки. Хотя основная роль АРР в сетчатке – это все же осуществление межсинаптической нейропередачи [35].
О значении тау-белка. Тау-белок – важный компонент внутриклеточного транспорта биологических веществ. Он является связующим белком микротрубочек аксоплазматического тока. В определенных патологических условиях тау-белок может под воздействием реакции фосфорилирования образовывать нерастворимые нитевидные агреганты, которые являются основным компонентом нейрофибриллярных клубочков (NFTs). Кодируется тау-белок геном тау-белка для стабилизации микротрубочек (МАРТ) в хромосоме 17 в положении 17q21. Экспрессия тау-белка происходит как в высших отделах головного мозга, так и в аксонах нейронов [47].
Основной функцией тау-белка является стабилизация микротрубочек [67]. Однако он обладает свойствами универсального регулятора внутриклеточных взаимодействий в ЦНС. Благодаря способности разрывать связь в микротрубочках он останавливает процессы передачи патологического сигнала внутри нейрона [20]. Также тау-белок оказывает активное влияние на рост и развитие нейронов [40]. В эксперименте на мышах с выключением экспрессии тау-белка резко снижался нейрогенез [37].
В сетчатке тау-белок не только поддерживает цитоскелет и регулирует аксональный транспорт в нейронах, но и влияет на накопление бета-амилоида и сигнализацию к устойчивости нервных клеток [50, 51, 57].
Зрительные нарушения у больных с болезнью Альцгеймера. Начиная с 1987 г. в литературе описываются различные изменения зрительных функций у больных с болезнью Альцгеймера [15]. Известно, что при болезни Альцгеймера нейродегенеративным изменениям подвергаются холинэргические нейроны и страдают в первую очередь когнитивные функции. При этом у них часто возникают трудности при чтении и поиске предметов [42, 44, 46]. У таких больных констатировались нарушения восприятия глубины, структуры и цвета предметов [17, 24, 55]. Долгое время эти зрительные нарушения объясняли повреждением нейронов зрительного пути [49]. Однако со временем появились доказательства того, что при болезни Альцгеймера изменения происходят не только в ЦНС, но и в самой сетчатке [70]. При помощи оптической когерентной томографии сетчатки у пациентов с болезнью Альцгеймера было выявлено значительное уменьшение толщины сетчатки в парапапиллярной зоне по сравнению с контрольной группой [5, 12, 39, 60, 62]. Также у пациентов с болезнью Альцгеймера наблюдали изменения зрительного нерва. При помощи конфокальной сканирующей лазерной офтальмоскопии были выявлены уменьшение толщины слоя нервных волокон сетчатки, объема нейроретинального ободка и увеличение отношения экскавации к диску зрительного нерва [19]. Эти данные были подтверждены при патологоанатомическом исследовании глаз. При гистопатологическом анализе сетчатки у пациентов с болезнью Альцгеймера выявляли уменьшение толщины слоя нервных волокон, количества крупных ганглионарных клеток и диаметра ДЗН [34, 65].
Также у пациентов с болезнью Альцгеймера описана высокая частота встречаемости ПОУГ [11, 68] – у них, по данным разных авторов, заболеваемость глаукомой в 2,5–5 раз больше, чем в общей популяции. Примечательно, что при болезни Альцгеймера, так же как и при глаукоме, происходит гибель ганглионарных клеток сетчатки и аксонов зрительного нерва по пути апоптоза [10]. Кроме того, наблюдаются схожие дефекты поля зрения и показатели ЭРГ [22, 58, 61, 64]. Таким образом, возможно, что в механизмах нейродегенерации при глаукоме, как и при болезни Альцгеймера определенную роль играют бета-амилоид и тау-белок.
Бета-амилоид и глаукома. В последние годы исследователи уделяют большое внимание роли бета-амилоида в развитии глаукомной нейрооптикопатии [74, 75]. Так, при моделировании офтальмогипертензии у мышей было выявлено повышенное количество белка – предшественника бета-амилоида у более взрослых мышей по сравнению с молодыми и интактными животными [26]. Другими авторами также было выявлено присутствие бета-амилоида у мышей с экспериментальной глаукомой, при этом уровень его носил дозозависимый эффект [54]. Введение веществ, блокирующих бета-амилоид (ингибитор бета-секретазы, антитела к анти-бета-амилоиду, антиагрегант бета-амилоида «Конго красный»), оказывало нейропротективный эффект при экспериментальной глаукоме [27]. Также на культуре выделенных ганглионарных клеток сетчатки мышей было продемонстрировано нейротоксическое действие предшественника бета-амилоида в присутствии индуктора синтеза бета-амилоида. Дегенерация нейронов сетчатки была пропорциональна времени воздействия БА и его концентрации. При добавлении в культуру клеток ингибитора синтеза бета-амилоида гибель клеток прекращалась [69].
Сывороточный амилоид при помощи ПЦР-реакции был также обнаружен у пациентов с глаукомой в трабекулярной зоне, а его уровень в сыворотке крови был выше, чем у пациентов без глаукомы [71]. Существует мнение, что прогрессирование глаукомы напрямую связано с уровнем бета-амилоида и тау-белка в спинномозговой жидкости у больных ПОУГ [59]. В настоящее время активно рассматривается возможность апробации препаратов, применяемых в лечении нейродегенеративных расстройств при болезни Альцгеймера, в терапии глаукомной нейрооптикопатии [8, 43].
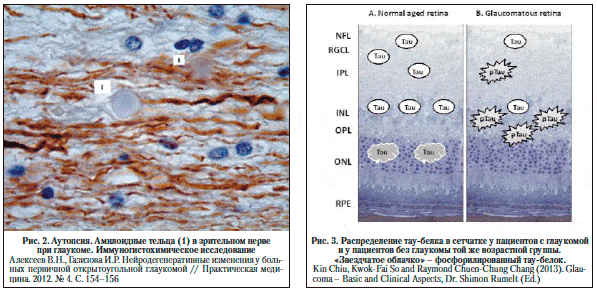
Особый интерес представляет результаты патологоанатомического исследования у людей с ПОУГ [1]. В обоих случаях у умерших пациентов, при жизни страдавших ПОУГ, выявлены процессы нейродегенерации. В дегенеративный процесс были вовлечены все уровни центрального отдела зрительного анализатора, но наиболее заметно – область зрительной коры в районе шпорной борозды. Особо следует выделить тот факт, что в зрительном нерве и в IV–V слоях коры головного мозга были обнаружены амилоидные бляшки и тельца (рис. 1).
Митохондриальная дисфункция и бета-амилоид. Нейротоксичный эффект бета-амилоида напрямую связан с его способностью открывать митохондриальные поры [8]. Механизм активации апоптоза по митохондриальному пути бета-амилоидом связывают с нарушением баланса Са2+. При этом нарушается гомеостаз кальция в цитозоле нервной клетки, митохондрии набухают, высвобождаются активаторы каспаз, запускается механизм необратимой гибели клетки по пути апоптоза [38, 52, 66]. Апоптоз – это основной механизм гибели клетки при нейродегенеративных заболеваниях. Сигналы от поврежденных участков клетки сходятся на митохондриях, вызывая повышение проницаемости обеих мембран, снижение мембранного потенциала и высвобождение белков апоптоза – апоптоз-индуцирующего фактора (AIF), SMAC (second mitochondria-derived activator of caspases) и некоторых прокаспаз – из межмембранного пространства. При поступлении апоптотического сигнала происходит транслокация AIF из митохондрии в цитоплазму, а затем в ядро.
Клинические исследования показали, что средняя активность митохондриальной функции была снижена на 21% у пациентов с ПОУГ по сравнению с людьми соответствующего возраста (р <0,001) [9].
Тау-белок и глаукома. При иммуногистохимическом исследовании ткани сетчатки у людей в возрасте от 49 до 87 лет была обнаружена прямая корреляция между количеством тау-позитивных ганглионарных клеток сетчатки и возрастом. Также диффузное расположение нефосфорилированного тау-белка было обнаружено во внутреннем ядерном слое сетчатки, а тау-белок в агрегатном состоянии был обнаружен в цитоплазме фоторецепторов. При этом тау-белок в сетчатке присутствовал у всех людей старше 63 лет [48]. По данным исследователей, тау-белок определяется и в сетчатке больных глаукомой, но в гораздо меньшем количестве. Однако при исследовании с моноклональными антителами AT8 выявления фосфорилированного тау-белка у больных глаукомой был выявлен гиперфосфорилированный тау-белок, который играет значимую роль в патогенезе болезни Альцгеймера. Локализован он был больше во внутреннем плексиформном слое, а также в ганглионарных волокнах сетчатки [28]. Распределение тау-белка в сетчатке у пациентов с глаукомой и у пациентов без глаукомы той же возрастной группы показано на рисунке 2. Повышенное содержание фосфорилированного тау-белка у пациентов с глаукомой указывает на подтверждение гипотезы, что прогрессирование нейродегенерации при глаукоме происходит по примерно такому же пути, как и при болезни Альцгеймера. Этим также объясняется факт большой встречаемости глаукомы у пациентов с болезнью Альцгеймера. Авторы также указывают на то, что изменение динамики движения и давления спинномозговой жидкости может влиять на транспортировку тау-белка и бета-амилоида в супрахориоидальное пространство и на прогрессирование глаукомы [28].
Таким образом, в представленном обзоре литературных данных мы попытались привести доказательную базу того, что прогрессирование нейродегенеративных изменений при глаукоме может быть обусловлено патологическим бета-амилоидом и гиперфосфорилированным тау-белком. При этом остается нерешенным вопрос о направлении прогрессирования нейродегенерации в проводящих путях зрительного анализатора при ПОУГ.

Литература
1. Алексеев В.Н., Газизова И.Р. Нейродегенеративные изменения у больных первичной открытоугольной глаукомой // Практическая медицина. 2012. № 4. С. 154–156.
2. Алексеев В.Н., Газизова И.Р., Никитин Д.Н. Морфологические особенности центральных отделов зрительного анализатора при экспериментальной глаукоме // В сб. науч. трудов по материалам конференции «Современные технологии диагностики и лечения при поражениях органа зрения». Спб.: ВМедА, 2013. С. 13–14.
3. Волков В.В. Глаукома при псевдонормальном давлении: Руководство для врачей. М.: Медицина, 2001. 352 с.
4. Газизова И.Р., Алмайсам Р. Нейродегенеративные изменения в головном мозге при глаукоме // Клиническая офтальмология. 2012. № 3. С. 88–91.
5. Еричев В.П., Панюшкина Л.А., Фомин А.В. Оптическая когерентная томография сетчатки и зрительного нерва в диагностике болезни Альцгеймера // Глаукома. 2013. № 1. С. 5–11.
6. Еричев В.П., Туманов В.П., Панюшкина Л.А. Глаукома и нейродегенеративные заболевания // Глаукома. 2012. №1. С. 62–68.
7. Иллариошкин С.Н. Конформационные болезни мозга. М.: Янус-К, 2003. 248 с.
8. Нестеров А.П. Глаукома. М.: Медицина, 2008. 360 с.
9. Abu-Amero K.K., Morales J., Bosley T.M. Mitochondrial abnormalities in patients with primary open-angle glaucoma // Invest. Ophthalmol. Vis. Sci. 2006. Vol. 47. Nо 6. P. 2533–2541.
10. Bayer A.U., Keller O.N., Ferrari F., Maag K.P. Association of glaucoma with neurodegenerative diseases with apoptotic cell death: Alzheimer's disease and Parkinson's disease // Am. J. Ophthalmol. 2002. № 1. Р. 135–137.
11. Bayer A.U., Ferrari F., Erb C. High occurrence rate of glaucoma among patients with Alzheimer's disease // Eur. Neurol. 2002. Vol. 47. Р. 165–168.
12. Berisha F., Feke G.T., Trempe C.L. et al. Retinal abnormalities in early Alzheimer's disease // Invest. Ophthalmol. & vis. Sci. 2007. Vol. 48. Р. 2285–2289.
13. Bizrah M., Guo L., Cordeiro M.F. Glaucoma and Alzheimer's disease in the elderly // Aging Health. 2011. №5. Р. 719–733.
14. Chow V.W., Mattson M.P., Wong P.C., Gleichmann M. An overview of APP processingenzymes and products // Neuromolecular. Med. 2010. Vol. 12. Р. 1–12.
15. Cogan D.G. Alzheimer syndromes // Am. J. Ophthalmol. 1987. Vol. 104. Р. 183–184.
16. Crawford M.L., Harwerth R.S., Smith E.L., et al. Glaucoma in primates: cytochrome oxidase reactivity in parvo- and magnocellular pathways // Invest. Ophthal. Vis. Sci. 2000. № 41. Р. 1791–1802.
17. Cronin-Golomb A. Vision in Alzheimer's disease // Gerontologist. 1995. Vol. 35. Р. 370–376.
18. Dai H., Mu K.T., Qi J.P. et al. Assessment of Lateral Geniculate Nucleus Atrophy with 3T MR Imaging and Correlation with Clinical Stage of Glaucoma // Am. J. Neuroradiol. 2011. № 32. Р. 1347–1353.
19. Danesh-Meyer H.V., Birch H., Ku J.Y. et al. Reduction of optic nerve fibers in patients with Alzheimer disease dentified by laser imaging // Neurol. 2006. Vol. 67. Р. 1852–1854.
20. Dawson H.N., Ferreira A., Eyster M.V. et al. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice // J. Cell Sci. 2001. Vol. 114. Р. 1179–1187.
21. Dinet V., An N., Ciccotosto G.D. et al. APP involvement in retinogenesis of mice // Acta Neuropathol. 2011. Vol. 121. Р. 351–363.
22. Foster P.J., Buhrmann R., Quigley H.A., Johnson G.J. The definition and classification of glaucoma in prevalence surveys // Bri. J. Ophthalmol. 2002. Vol. 86. Р. 238–242.
23. Garaci F.G., Bolacchi F., Cerulli A. et al. Optic Nerve and Optic Radiation Neurodegeneration in Patients with Glaucoma: In Vivo Analysis with 3-T Diffusion-Tensor MR Imaging // Radiol. 2009. № 2. Р. 496–501.
24. Gilmore G.C., Whitehouse P.J. Contrast sensitivity in Alzheimer's disease: a 1-year longitudinal analysis // Optom. Vis. Sci. 1995. Vol. 72. Р. 83–91.
25. Goate A., Chartier-Harlin M.C., Mullan M. et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease // Nature. 1991. Vol. 349. Р. 704–706.
26. Goldblum D., Kipfer-Kauer A., Sarra G.M. et al. Distribution of amyloid precursor protein and amyloid-beta immunoreactivity in DBA/2J glaucomatous mouse retinas // Inves. Ophthalmol. & Vis. Sci. 2007. № 11. Р. 5085–5090.
27. Guo L., Salt Th.E., Luong V., Wood N. Targeting amyloid-b in glaucoma treatment // PNAS. 2007. №14. Р. 13444–13449.
28. Gupta N., Fong J., Ang L.C., Yucel Y.H. Retinal tau pathology in human glaucomas // Can. J. Ophthalmol. 2008. Vol. 43. Р. 53–60.
29. Gupta N., Ang L.C., Noel de Tilly L. et al. Human glaucoma and neural degeneration in intracranial optic nerve, lateral geniculate nucleus and visual cortex // Br. J. Ohthalmol. 2006. Vol. 90. P. 674–678.
30. Gupta N., Greenberg G., de Tilly L.N. et al. Atrophy of the lateral geniculate nucleus in human glaucoma by magnetic resonance imaging // Br. J. Ohthalmol. 2008. Vol. 93. P. 56–60.
31. Gupta N., Yucel Y.H. Glaucoma and the brain // J. Glaucoma. 2001. № 10. Р. 28–29.
32. Gupta N., Yucel Y.H. Glaucoma as a neurodegenerative disease // Curr. Opin. Ophthalmol. 2007. № 2. Р. 110–114.
33. Herard A.S., Besret L., Dubois A. et al. siRNA targeted against amyloid precursor protein impairs synaptic activity in vivo // Neurobiol. Aging. 2006.- Vol. 27. Р. 1740–1750.
34. Hinton D.R., Sadun A.A., Blanks J.C., Miller C.A. Optic-nerve degeneration in Alzheimer's disease // N. Engl. J. Med. 1986. Vol. 315. Р. 485–487.
35. Ho T., Vessey K.A., Cappai R. et al. Amyloid Precursor Protein Is Required for Normal Function of the Rod and Cone Pathways n the Mouse Retina // PLoS One. 2012. № 7.
36. Hoe H-S., Lee K.J., Carney R.S.E. et al. Interaction of Reelin with Amyloid Precursor Protein Promotes Neurite Outgrowth // J. Neurosci. 2009. Vol. 29. Р. 7459–7473.
37. Hong M., Zhukareva V., Vogelsberg-Ragaglia V. et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17 // Sci. 1998. Vol. 282. Р. 1914–1917.
38. Hung C.H., Ho Y.S., Chang R.C.C. Modulation of mitochondrial calcium as a pharmacological target for Alzheimer's disease // Ageing Res. Rev. 2010. Vol. 9. Р. 447–456.
39. Iseri P.K., Altinas O., Tokay T., Yuksel N. Relationship between cognitive impairment and retinal morphological and visual functional abnormalities in Alzheimer disease // J. Neuroophthalmol. 2006. Vol. 26. Р. 18–24.
40. I ttner L.M., Ke Y.D., Delerue F. et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models // Cell. 2010. Vol. 142. Р. 387–397.
41. Izzotti A., Sacca S.C., Longobardi M., Cartiglia C. Mitochondrial damage in the trabecular meshwork of patients with glaucoma // Arch. Opthalmol. 2010. Vol. 128. № 6. P. 724–730.
42. Jackson GR, Owsley C. Visual dysfunction, neurodegenerative diseases, and aging // Neurol Clin. 2003. Vol. 21. Р. 709–728.
43. Jiahua F., Fagang J., Jingbo L. et al. Rationale for the use of multifunctional drugs as neuroprotective agents for glaucoma // Neural Regenerat. Res. 2012. № 4. Р. 313–318.
44. Katz B., Rimmer S., Iragui V., Katzman R. Abnormal pattern electroretinogram in Alzheimer's disease: evidence for retinal ganglion cell degeneration? // Ann. Neurol. 1989. Vol. 26. Р. 221–225.
45. Chiu K., So K.-F., Chuen-Chung Chang R. Progressive Neurodegeneration of Retina in Alzheimer’s Disease — Are β-Amyloid Peptide and Tau New Pathological Factors in Glaucoma? // Glaucoma. Basic and Clinical Aspects. – 2013. Rumelt Sh. (Ed.), аvailable from: http://www.intechopen.com/books/glaucoma-basic-and-clinical-aspects/progressive-neurodegeneration-of-retina-in-alzheimer-s-disease-are-amyloid-peptide-and-tau-new-patho.
46. Lee A.G., Martin C.O. Neuro-ophthalmic findings in the visual variant of Alzheimer's disease // Ophthalmol. 2004. Vol. 111. P. 376–380.
47. Lee V.M-Y., Goedert M., Trojanowski J.Q. Neurodegenerative tauopathies // Annual Review of Neuroscience. 2001. Vol. 24.
P. 1121–1159.
48. Leger F., Fernagut P.O., Canron M.H. et al. Protein aggregation in the aging retina // J. Neuropath. Exp. Neurol. 2011. Vol. 70. P. 63–68.
49. Leuba G., Saini K. Pathology of subcortical visual centers in relation to cortical degeneration in Alzheimers-disease // Neuropath. Applied Neurobiol. 1995. Vol. 21. P. 410–422.
50. Lieven C.J., Millet L.E., Hoegger M.J., Levin L.A. Induction of axon and dendrite formation during early RGC-5 cell differentiation // Exp. Eye Res. 2007. Vol. 85. P. 678–683.
51. Liu B., Rasool S., Yang Z. et al. Amyloid-peptide vaccinations reduce {beta}-amyloid plaques but exacerbate vascular deposition and inflammation in the retina of Alzheimer's transgenic mice // The American journal of pathology. 2009. Vol. 175. P. 2099–2110.
52. Liu X., Hajnoczky G. Ca2+-dependent regulation of mitochondrial dynamics by the Miro-Milton complex // Int. J. Biochem. Cell. Biol. 2009. Vol. 41. P. 1972–1976.
53. McKinnon S.J. Glaucoma: ocular Alzheimer’s diseases? // Front biosci. 2003. Vol. 8. P. 1140–1156.
54. McKinnon S.J., Lehman D.M., Kerrigan-Baumrind L.A. et al. Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension // Invest. Ophthalmol. Visual. Sci. 2002. Vol. 43. P. 1077–1087.
55. Mendez M.F., Cherrier M.M., Meadows R.S. Depth perception in Alzheimer's disease // Percept. Mot. Skills. 1996. Vol. 83. P. 987–995.
56. Moya K.L., Benowitz L.I., Schneider G.E., Allinquant B. The amyloid precursor protein is developmentally regulated and correlated with synaptogenesis // Dev. Biol. 1994. Vol. 161.– P. 597–603.
57. Nakayama T., Goshima Y., Misu Y., Kato T. Role of cdk5 and tau phosphorylation in heterotrimeric G protein-mediated retinal growth cone collapse // J. Neurobiol. 1999. Vol. 41. P. 326–339.
58. Nesher R., Trick G.L. The pattern electroretinogram in retinal and optic-nerve disease - a quantitative comparison of the pattern of visual dysfunction // Doc. Ophthalmol. 1991. Vol. 77. P. 225–235.
59. Nucci C., Martucci A., Martorana A. et al. Glaucoma progression associated with altered cerebral spinal fluid levels of amyloid beta and tau proteins // Clin. & Experim. Ophthalmol. 2011. Vol. 3. P. 279–281.
60. Paquet C., Boissonnot M., Roger F., Dighiero P., Gil R., Hugon J. Abnormal retinal thickness in patients with mild cognitive mpairment and Alzheimer's disease // Neurosci. Lett. 2007. Vol. 420. P. 97–99. 172 Glaucoma - Basic and Clinical Aspects
61. Parisi V., Restuccia R., Fattapposta F. et al. Morphological and functional retinal impairment in Alzheimer's disease patients // Clin. Neurophysiol. 2001. Vol. 112. P. 1860–1867.
62. Parisi V. Correlation between morphological and functional retinal impairment in patients affected by ocular hypertension, glaucoma, demyelinating optic neuritis and Alzheimer's disease // Semin. Ophthalmol. 2003. Vol. 18. P. 50–57.
63. Priller C., Bauer T., Mitteregger G., Krebs B., Kretzschmar H.A., Herms J. Synapse formation and function is modulated by the amyloid precursor protein // J. Neurosci. 2006. Vol. 26. P. 7212–7221.
64. Quigley H.A. Glaucoma // Lancet. 2011. Vol. 377. P. 1367–1377.
65. Sadun A.A., Bassi C.J. Optic nerve damage in Alzheimer's disease // Ophthalmology. 1990. Vol. 97. P. 9–17.
66. Saotome M., Safiulina D., Szabadkai G. et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase // Proc. Natl. Acad. Sci. USA. 2008. Vol. 105. P. 20728–20733.
67. Spires-Jones T.L., Stoothoff W.H., de Calignon A., Jones P.B., Hyman B.T. Tau pathophysiology in neurodegeneration: a tangled ssue // Trends. Neurosci. 2009. Vol. 32. P. 150–159.
68. Tamura H., Kawakami H., Kanamoto T. et al. High frequency of open-angle glaucoma in Japanese patients with Alzheimer's disease // J. Neurol. Sci. 2006. Vol. 246. P. 79–83.
69. Tsuruma K., Tanaka Y., Shimazawa M., Hara H. Induction of amyloid precursor protein by the neurotoxic peptide, amyloid-beta 25-35, causes retinal ganglion cell death // J. of Neurochem. 2010. Vol. 6. Р. 1545–1554.
70. Valenti D.A. Neuroimaging of retinal nerve fiber layer in AD using optical coherence tomography // Neurology. 2007. Vol. 69. P. 1060.
71. Wang W.H., McNatt L.G., Pang I.H. et al. Increased expression of serum amyloid A in glaucoma and its effect on intraocular pressure // Inves. Ophthalmol. & Vis. Sci. 2008. Vol. 5. Р. 1916–1923.
72. Wang Z., Wang B., Yang L. et al. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis // J. Neurosci. 2009. Vol. 29. P. 10788–10801.
73. Yang L., Wang B., Long C. et al. Increased asynchronous release and aberrant calcium channel activation in amyloid precursor protein deficient neuromuscular synapses // Neurosci. 2007. –Vol. 149. P. 768–778.
74. Yin H., Chen L., Chen X., Liu X. Soluble amyloid beta oligomers may contribute to apoptosis of retinal ganglion cells in glaucoma // Med. Hypoth. 2008. Vol. 1. P. 77–80.
75. Yoneda S., Hara H., Hirata A. et al. Vitreous fluid levels of beta-amyloid(1-42) and tau in patients with retinal diseases // Jpn. J. Ophthalmol. 2005. Vol. 2. P. 106–108.



