




Семейные доброкачественные гипербилирубинемии (идиопатические негемолитические гипербилирубинемии, доброкачественные гипербилирубинемии, пигментные гепатозы) – группа заболеваний без выраженного изменения структуры и функции печени и без явных признаков гемолиза и холестаза, обусловлены нарушением обмена билирубина (Е 80 по МКБ–10), что проявляется стойкой или перемежающейся желтухой [1]. Идиопатические негемолитические гипербилирубинемии составляют большую группу состояний, развитие которых обусловлено нарушением внутриклеточного транспорта билирубина. При этом нет признаков гемолиза или заболеваний печени. Большинство из них (важнейшее исключение – синдром Криглера–Найяра) доброкачественны. Эти гипербилирубинемии возникают как за счет непрямого, так и прямого билирубина.
К непрямым (неконъюгированным) гипербилирубинемиям относятся синдромы Жильбера (Е 80.4), Криглера–Найяра (Е 80.5), Дрискола, первичная семейная гипербилирубинемия. К прямым (конъюгированным) гипербилирубинемиям относятся синдромы Дабина–Джонса и Ротора.
Наиболее распространенным состоянием в этой группе заболеваний является синдром Жильбера (СЖ). СЖ наблюдается у 3–7% европейцев, 3% азиатов и 36% африканцев. Среди носителей признака мужчин в 2–7 раз больше, чем женщин. Несмотря на широкую распространенность СЖ в популяции, этот синдром далеко не всегда учитывается при проведении дифференциальной диагностики (ДД) желтух, а в случае его выявления возникает много вопросов по определению врачебной тактики [2].
СЖ наследуется аутосомно–рецессивно (а/р), характеризуется интермиттирующей желтухой без признаков гемолиза или болезней печени. Гипербилирубинемия умеренная (не выше 6 мг%, у большинства – менее 3 мг%), варьирует по дням недели, времени года, наличия или отсутствия обезвоживания, голодания, физических нагрузок, интеркурентных заболеваний, менструации. Нередко каких–либо объяснений появлению желтушности найти не удается. Эпизоды желтушности завершаются самостоятельно. Повышение билирубина не отмечается более чем у 1/3 носителей гена.
Патогенез
Неконъюгированная гипербилирубинемия при СЖ обусловлена снижением активности энзимов системы билирубин–уридин дифосфат глюкоронил трансферазы (БУДГТ). БУДГТ находится в эндоплазматическом ретикулуме гепатоцитов и осуществляет перевод билирубина в билирубин моноглюкоронид и диглюкорогид. БУДГТ – только одна из изоформ уридин глюкоронил трансфераз (УГТ), ответственных за конъюгацию гормонов, нейротрансмиттеров, карциногенов и многих других соединений.
Ген уридин глюкоронил транферазы расположен на 2–й хромосоме, содержит 5 экзонов. Экзоны 2–5 являются постоянными компонентами всех изоформ УГТ. Экзон 1 кодирует специфические регионы энзима и имеет базовые основания ТАТАА. Экзоны 1а и 1d кодируют регионы БУДГТ 1А1 и БУДГТ 1А2 соответственно. БУДГТ 1А1 отвечает за полную конъюгацию билирубина [3]. Метаболическое значение БУДГТ 1А2 невелико. Экспрессия БУДГТ 1А1 зависит от промотера в позиции 5’. Таким образом, нарушение глюкоронирования билирубина зависит от мутации в экзоне 1А, его промотере или в общем экзоне. Мутация в экзоне заключается в появлении двух дополнительных оснований (ТА). Добавление новых оснований к прежним ТАТАА нарушает взаимодействие с фактором транскрипции IID, экспрессия БУДГТ снижается на 30%. В гомозиготных случаях в желчи над диглюкоронидом преобладает моноглюкоронид билирубина.
В промотерном регионе описаны дополнительные мутации (Gly71Arg, Pro364Leu, Tyr468Asp). У носителей этих мутаций концентрация билирубина в крови существенно превышает нормальные показатели.
Степень гипербилирубинемии и клинические проявления СЖ зависят не только от снижения активности БУДГТ, но и от сопутствующих факторов: скрытого гемолиза, нарушения внутрипеченочного транспорта. Например, у многих людей с дефектом ТАТАА нет неконъюгированной гипербилирубинемии, точно так же, как у пациентов с гранулематозными заболеваниями печени и снижением активности БУДГТ.
Клиническая картина
Клиническая картина СЖ вариабельна. Синдром нередко ассоциируется с генерализованной дисплазией соединительной ткани по типу синдромов Марфана или Элерса–Данлоса. У детей грудного возраста, гомозиготных по СЖ и находящихся на грудном вскармливании, желтуха новорожденных выражена ярче и длится дольше, чем у детей без СЖ [4]. Обычно СЖ выявляется в период пубертата. Видимо, манифестирующая иктеричность провоцируется дополнительным угнетением глюкоронирования билирубина половыми гормонами. В более старшем возрасте СЖ выявляется в период интеркурентных заболеваний. В этом случае приходится проводить ДД с постгепатической гипербилирубинемией. Расспрос больного о желтухах у родственников может оказать помощь в установлении диагноза.
Многие пациенты эмоциональны, отмечают гиперчувствительность кожных покровов. При обследовании выявляются умеренная желтуха (чаще иктеричность склер на фоне матово–желтушной кожи, особенно лица). Иногда наблюдается частичное окрашивание носогубного треугольника, ладоней, подмышечных впадин, стоп. Желтуха усиливается при дегидратации, голодании, инфекции (в том числе вирусной), приеме алкоголя, тяжелой физической и умственной работе, стрессах. Описывают пылающие и пигментные невусы, пигментацию век, астенический синдром (депрессия, неспособность концентрировать внимание, повышенная утомляемость, слабость, плохой сон и т.д.). Очень часто больные предъявляют жалобы на боли в животе. Абдоминальный синдром чаще мультифакториальный, нередко ассоциируется с общей тревожностью. Корреляций между наличием или выраженностью абдоминального синдрома и степенью гипербилирубинемии нет. У 50% больных диагностируется скрытый гемолиз (группа риска по холелитиазу!) (рис. 1). Камни в желчном пузыре, по нашему опыту, у таких больных могут сформироваться очень быстро.
Размеры печени и селезенки остаются, как правило, нормальными. Билирубинурия отсутствует. Исследование печеночных ферментов решающего диагностического значения не имеет, хотя в редких случаях описано повышение сывороточной щелочной фосфатазы. Как и при других видах доброкачественных гипербилирубинемий, гистологическое строение печени близко к норме. Признаков диспротеиноза, некроза печеночных клеток обычно не обнаруживается. Но вокруг терминальных печеночных венул обнаруживается накопление липофусцин подобного пигмента.
Лабораторная и дифференциальная диагностика синдрома Жильбера
Лабораторная диагностика СЖ основана на биохимических исследованиях. Генетические исследования дорогостоящи и недоступны в широкой практике.
Проба с голоданием. Резкое ограничение энергетической ценности пищи в течение 48 часов вызывает повышение уровня неконъюгированного билирубина. В течение 48 часов больной получает питание энергетической ценностью 400 ккал/сут. В день начала пробы утром натощак и спустя двое суток определяют билирубин сыворотки крови. При подъеме его на 50–100% проба считается положительной. Через 24 часа после возобновления нормального питания уровень неконъюгированного билирубина возвращается к норме. Причины повышения концентрации неконъюгированного билирубина при голодании пока не установлены. Концентрация неконъюгированного билирубина на фоне голодания повышается и у пациентов с гемолитической анемией или заболеваниями печени, но степень повышения значительно меньше. Повышение концентрации билирубина наблюдается и при нормокалорийной диете, но при ограничении жиров. После приема жиров концентрация билирубина быстро возвращается к норме. В современных условиях проба с голоданием используется редко.
Никотиновая кислота в дозе 50 мг при в/в введении в ближайшие 3 часа приводит к 2–3–кратному повышению концентрации неконъюгированного билирубина. Механизм действия сводится к осмотическому разрушению эритроцитов, повышенному образованию билирубина в селезенке, обратимой ингибиции БУДГТ. Специфичность пробы недостаточно высока. Аналогичная реакция может быть при гемолитических анемиях и при заболеваниях печени.
Фенобарбитал и другие индукторы БУДГТ нормализуют концентрацию билирубина в плазме.
Тонкослойная хроматография позволяет выявить повышение концентрации моноглюкоронида билирубина по отношению к диглюкорониду, что свойственно СЖ.
Клиренс препаратов, определяемый по бромсульфофталеиновой пробе, индоцианину зеленому снижен.
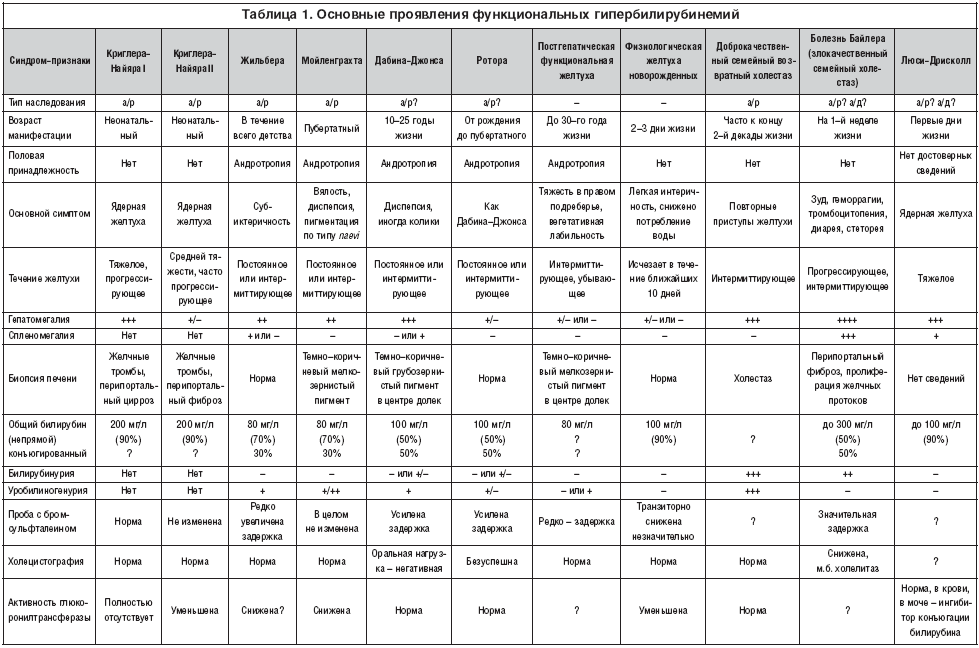
Дифференциальную диагностику проводят с другими вариантами семейных гипербилирубинемий (табл. 1).
Синдром Криглера–Найяра = семейная негемолитическая желтуха новорожденных. Впервые описана в 1952 г. Некоторые считают, что синдром Криглера–Найяра – вариант более тяжелых мутаций в том же сегменте ТАТАА, как и у СЖ. Это объясняет промежуточные концентрации билирубина у родственников больных синдромом Криглера–Найяра. Различают две генетически гетерогенные формы болезни.
При первой форме синдром наследуется а/р. Обусловлен практически полным отсутствием БУДГТ. Интенсивная, часто ядерная желтуха обусловлена 20–30–кратным повышением непрямого билирубина сыворотки крови. Развивается в первые часы и дни жизни ребенка. На первый план выступают симптомы поражения ЦНС: мышечная гипотония, нистагм, опистотонус, атетоз, тонические и клонические судороги, отставание в физическом и умственном развитии. Гематологические показатели остаются в пределах нормы. Билирубинурия отсутствует, количество уробилиновых тел в моче и кале невелико. Успеха от применения фенобарбитала или глутетимида, индукторов микросомальных ферментов, нет. Больные редко доживают до 1,5 лет.
При второй форме синдром наследуется а/д. БУДГТ присутствует, хотя активность фермента значительно снижена. Интенсивность желтухи менее выражена. Ядерная желтуха не развивается. Эффект фототерапии, индукторов микросомальных ферментов хороший. Уровень непрямой фракции билирубина сыворотки крови повышается в 5–20 раз. Желчь окрашена, в кале определяется большое количество уробилиногена. Больные доживают до 50 лет и более, но в отдаленном периоде, особенно при позднем лечении, нередки глухота, хореатетоз, нейромышечные и личностные отклонения, гипоплазия зубов.
Синдром Дрискола – семейная форма преходящей гипербилирубинемии новорожденных за счет стероидных тел в плазме матери, блокирующих конъюгацию билирубина. Близкое (но не семейное) состояние известно у некоторых детей при вскармливании грудным молоком, в котором может содержаться не уточненный фактор, препятствующий глюкуронированию билирубина.
Синдром Дабина–Джонсона. Описан в 1954 г. Передается а/р. Проявляется хронической негемолитической желтухой с повышением уровня связанного билирубина в сыворотке крови. Часто начинается в период полового созревания, но может начаться в любом возрасте. Желтуха носит хронический или интерметтирующий характер, содержание билирубина не превышает 0,06 г/л. Обострение желтухи проявляется болями в правом подреберье, общей слабостью, наблюдается при наслоении инфекций. Во время ремиссии желтуха почти полностью исчезает. Печень умеренно увеличена, плотноватой консистенции. В половине всех случаев пальпируется селезенка. Активность ферментов сыворотки крови и осадочные функциональные пробы печени остаются нормальными. В моче обнаруживаются желчные пигменты. Признаки гемолиза отсутствуют. Бромсульфалеиновая проба изменена. При назначении бромсульфалеина его содержание в крови быстро падает, чтобы затем вновь подняться; на 90–й и 120–й минуте концентрация превышает таковую на 45–й минуте, то есть происходит нормальный захват красителя с затруднением освобождения из клеток печени. Достоверно диагноз можно установить при пункционной биопсии, выявляющей отложение в клетках печени крупнозернистого меланиноподобного пигмента темно–коричневого цвета. При лапароскопии печень зеленовато–коричневая, желчный пузырь не изменен.
Вариантом синдрома Дабина–Джонсона (или самостоятельной единицей?) является синдром Бюрка. При нем также обнаруживается липохромный гепатоз, но БЕЗ желтухи, хотя и со значительной гепатоспленомегалией.
Синдром Ротора описан в 1948 г. Передается а/р. Протекает с преимущественным повышением уровня связанного билирубина в сыворотке крови при нормальной гистологической картине печени. Болезнь протекает, как длительная желтуха с умеренным повышением конъюгированного билирубина при нормальных показателях функции печени и хорошим контрастированием желчного пузыря при холецистографии. ДД с синдромом Дабина–Джонсона проводится на основании пункционной биопсии, поскольку при синдроме Ротора никогда не обнаруживается темного пигмента в клетках печени.
Наряду с этими синдромами ДД синдрома Жильбера проводят с общим синдромом желтух, в том числе с гемолизом, гематомами, острыми и хроническими заболеваниями печени, рабдомиолизом, неэффективным эритропоэзом, побочным действием препаратов.
Лечение синдрома Жильбера
Пациента следует информировать, что СЖ является доброкачественной гипербилирубинемией, продолжительность жизни в среднем не отличается от общей популяции. Тем не менее могут возникнуть холелитиаз или вышеуказанные соматические и психосоматические расстройства, что требует коррекции режима и в ряде случаев медикаментозного вмешательства.
Госпитализаций при неосложненном течении не требуется. Прививочный календарь не изменяется. Физическая нагрузка должна быть обычной. Не следует делать больших перерывов в еде. Неблагоприятно ограничение жидкости.
Фенобарбитал или глютетемид индуцируют активность БУДГТ и могут использоваться короткими курсами при неблагоприятных ситуациях [5].
Учитывая, что при СЖ наряду с биохимическими нарушениями страдает захват молекул гепатоцитами и транспорт по клетке, существует риск желчно–каменной болезни, мы применили у 15 наших больных препарат ЛИВ.52К.
ЛИВ.52К широко используется в гастроэнтерологии и гепатологии, в педиатрии (у детей старше 2 лет по 10–20 капель 2 раза в сутки) при хронических и острых гепатитах, дисфункциях желчевыводящей системы и многих других состояниях. ЛИВ.52К представляет собой комбинированный растительный препарат. Препарат выпускается в таблетках и каплях, легко переносится детьми, оказывает гепатопротекторное, антитоксическое, противовоспалительное, желчегонное, антиоксидантное, антианорексическое действие. Гепатопротекторное действие ЛИВ.52К обусловлено антиоксидантными и мембраностабилизирующими свойствами входящих в его состав компонентов. ЛИВ.52К стимулирует биосинтез белков и фосфолипидов, способствует восстановлению гепатоцитов, уменьшает дегенеративные, жировые и фиброзные изменения, усиливает внутриклеточный обмен. Препарат регулирует уровень плазменных белков крови, нормализуя соотношение альбумин/глобулин. Обеспечивает нормализацию уровня плазменных трансаминаз, холестерина, триглицеридов, уменьшая проявления дислипидемии. Снижает показатели билирубина и щелочной фосфатазы. Повышает способность печени к депонированию гликогена. Очень ценно, что препарат улучшает коллоидные свойства желчи, предупреждает образование желчных камней и способствует нормализации сократительной функции желчного пузыря.
На фоне применения препарата улучшалось общее состояние детей. Нормализовались аппетит, исчезала гиперестезия кожных покровов. В период вирусных инфекций нарушений билирубинового обмена не отмечено. В течение ближайших нескольких дней после начала приема ЛИВ.52К исчезали боли в правом подреберье. По данным ультразвуковых исследований исчезал феномен сладжа в желчном пузыре.
Литература
1. Siegenthaler W. (Hrsg.) Differentialdiagnose innerer Krankheiten. – 17. neubearbeitete Auflage. – Stuttgart: Georg Thieme Verlag, 1993.
2. Mukherjee S. Gilbert syndrome. eMedicine, Last Updated 15 May 2008. http://www.emedicine.com/med/TOPIC870.HTM
3. Ferraris A., D’Amato G., Nobili V. et al. Combined test for UGT1A1 –3279T—>G and A(TA)nTAA polymorphisms best predicts Gilbert’s syndrome in Italian pediatric patients. // Genet. Test, 2006. – v. 10. – pp. 121 – 125.
4. Bancroft J., Kreamer B., Gourley G. Gilbert syndrome accelerates development of neonatal jaundice. // Journal of Pediatrics, 1998. – v. 132. – pp. 656 – 660.
5. Black M., Sherlock S. Treatment of Gilbert’s syndrome with phenobarbitone. // Lancet, 1970. – v. 1. – pp. 1359 – 1361.



