




Пациентка Н., 34 лет поступила в пульмонологическое отделение Госпитальной терапевтической клиники ММА имени И.М. Сеченова в феврале 2010 г. с жалобами на общую слабость, боль в грудной клетке при глубоком вдохе, одышку при физической нагрузке (подъем на 2–й этаж), снижение толерантности к физической нагрузке, постоянные ноющие боли в околопупочной области живота.
Женщина считает себя больной с июня 2006 г., когда после полета в самолете из Москвы в Ташкент резко начала нарастать одышка, в течение 2 нед. постепенно появился дискомфорт слева в кардиальной области. Больная спала только на левом боку (на правом начинала сразу задыхаться), появился сухой кашель. Одышка стала беспокоить даже в покое. Она обратилась в больницу по месту своего пребывания, где при рентгенографии органов грудной клетки был определен тотальный пневмоторакс слева, в связи с чем была выполнена торакоскопическая электрокоагуляция буллы левого легкого. После операции женщина чувствовала себя удовлетворительно. Через 3 нед., в июле 2006 г., вновь отметила нарастание вышеуказанных жалоб – постепенно увеличилась одышка, теперь Н. могла спать только на правом боку, кашель с выделением слизистой мокроты. Пациентка была повторно госпитализирована, выполнена торакоскопическая электрокоагуляция булл правого легкого, плевродез в связи с развившимся правосторонним пневмотораксом. После операции и в течение 2007–2008 гг. чувствовала себя удовлетворительно, но продолжала беспокоить общая слабость. С начала 2009 г. вновь появилась боль на вдохе в левой половине грудной клетки, постепенно появилась одышка при умеренной физической нагрузке (подъем на 2–й этаж).
На проведенной рентгенографии патологических изменений не обнаружилось, и в течение последующего года пациентка к специалистам не обращалась.
С начала февраля 2010 г. помимо вышеуказанных жалоб больная отметила появление дистанционных хрипов при дыхании и постоянных ноющих болей в околопупочной области и левой половине живота. Обратилась за консультацией к пульмонологу Клиники госпитальной терапии ММА имени И.М. Сеченова. Была проведена компьютерная томография органов грудной клетки: в обоих легких определялись множественные кисты размерами от 4 до 36 мм; субплеврально – множественные буллы размером до 43 мм; справа в средней доле – множественные крупные кисты и участки фиброза.
В связи с выявленными изменениями в легких пациентка была госпитализирована в пульмонологическое отделение ГТК для уточнения диагноза.
В анамнезе жизни пациентки обращало внимание наличие частых инфекций НДП и ОРВИ. Женщина не курит и никогда не курила. Наследственность не изменена. Гинекологический анамнез пациентки – менструации с 14 лет, до 30–летнего возраста были нерегулярными (цикл – от 28 дней до полугода), умеренными, по 7 дней. С 15 лет ставился диагноз хронического аднексита. Беременностей не было, лечения не проводилось. Уровень половых гормонов в норме. Диагностировано первичное бесплодие. В 30–летнем возрасте (2005 г.) – выявили полип в полости матки, поликистоз яичников. Со слов больной, в анализах крови определялось увеличение уровня тестостерона. Было произведено выскабливание стенок полости матки, выправление загиба левой маточной трубы, резекция кист обоих яичников. Проведено гистологическое исследование (со слов пациентки – патологические изменения не выявлены, документации нет). В 2008 г. обнаружен полип шейки матки, не лечилась. С января 2010 г. – менструации стали обильными, вне menses стали возникать обильные кровянистые выделения алой кровью, впервые появились боли в левой паховой области.
При объективном осмотре обращали на себя внимание: ослабленное дыхание, с жестким оттенком и единичными сухими хрипами при форсированном дыхании, а также болезненность при пальпации живота в околопупочной области и левой половине живота, здесь же определялось образование плотноэластической консистенции размерами 10х8 см. Дизурии не отмечала. По результатам проведенного обследования, патологии в анализах крови и мочи не выявилось. На ЭКГ – синдром S1–S2–S3. Перегрузка правого предсердия. Проведено исследование функции внешнего дыхания: ЖЕЛ 2,39 л (73,15%), ОФВ1 1,75 л (62,23%), индекс Тиффно 73,55%. Умеренные вентиляционные нарушения по смешанному типу, умеренно выраженная генерализованная обструкция.
Результаты повторной МСКТ органов грудной клетки (рис. 1): в обоих легких определялись множественные кисты размерами от 4 до 36 мм. Субплеврально – множественные буллы размером 43 мм. Справа в средней доле – множественные крупные кисты и участки фиброза. Плотность легочной паренхимы несколько повышена. В 6–м и 9–м сегментах правого легкого определялись участки фиброза, плевральные сращения. Плевра неравномерно утолщена. Жидкости в плевральных полостях нет. В плевральной полости справа имеется небольшое количество воздуха. Осумкованный воздух определяется также в междолевых щелях с обеих сторон. Трахея и крупные бронхи свободно проходимы, не деформированы. Лимфатические узлы средостения не увеличены. Аорта, легочный ствол и их ветви не расширены. Листки перикарда тонкие, жидкости в полости перикарда нет.
При УЗИ органов брюшной полости и почек были выявлены: полиповидный холестероз желчного пузыря, диффузные изменения паренхимы печени и поджелудочной железы, объемные образования брюшной полости (возможно, исходящие из корня брыжейки или кистома левого яичника): печень в передне–заднем размере не увеличена: правая доля 136 мм, левая – 62 мм, паренхима диффузная, изменена по типу жировой дистрофии. Сосудистый рисунок печеночных вен не изменен. Внутрипеченочные желчные протоки не расширены. Желчный пузырь 69х25 мм, неправильной формы, в теле ближе к шейке перегиб, стенки уплотнены, с множественными пристеночными образованиями до 6 мм, камней нет. Общий желчный проток 4,6 мм. Воротная вена 11 мм. Поджелудочная железа не увеличена, уплотнена, главный панкреатический проток не расширен. Селезенка 107х41 мм. Свободной жидкости в брюшной полости нет. В мезогастральной и гипогастральной области слева визуализировалось гипоэхогенное образование с толстыми стенками 194х95 мм, с множественными перегородками, подвижное при дыхании. Подобное образование в малом тазу слева от матки размерами 85х62 мм. Почки обычно расположены, контуры ровные, толщина паренхимы до 18 мм. Патологических образований не выявлено, область надпочечников не изменена.
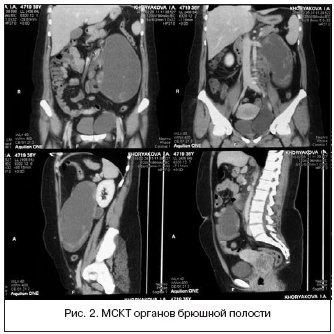
При МСКТ органов брюшной полости, забрюшинного пространства и таза (рис. 2) получена картина внеорганных образований брюшной полости, забрюшинного пространства и таза, диффузные изменения печени, косвенные признаки портальной гипертензии: в левой половине брюшной полости определялись внеорганное образование неправильной овальной формы, размерами 124х72х202 мм, пониженной плотности, с участками мягкотканной плотности по середине. На 10 мм выше нижней брыжеечной артерии слева от срединной линии визуализировались два тонких добавочных сосуда, кровоснабжающих опухоль, отходящих одним стволом. Мягкотканные участки активно накапливали контрастный препарат, преимущественно в паренхиматозную фазу, также определялись перегородки, накапливающие препарат. По периферии образования, парааортально, в области бифуркации аорты по ходу левых общих и наружных подвздошных сосудов визуализировались множественные подобные образования размером до 32 мм, с неровными контурами, по периферии накапливающие контрастный препарат идентично образованию (лимфоузлы?), подобное образование размерами 90х62 мм определялось в малом тазу, слева от матки.
Полученные результаты МСКТ органов грудной клетки и брюшной полости заставили нас исключить лимфангиомиоматоз.
Пациентка была переведена в клинику акушерства и гинекологии, где после проведенного обследования была обнаружена миома матки и произведена миомэктомия, по результатам гистологического исследования была получена картина лейомиомы матки (рис. 3).
Затем пациентка была переведена в Клинику факультетской хирургии им. Н.Н. Бурденко для дальнейшего хирургического лечения. Интраоперационно было получено: в забрюшинной клетчатке слева и справа от аорты имелись несколько ненапряженных жидкостных образований, проекционно исходящих из лимфатических протоков, связанных с парааортальными лимфоузлами и лимфоузлами корня брыжейки толстой кишки. Кисты размером от 5 до 20 см в диаметре в просвете содержали лимфу. Также определялось расширение лимфоидных капилляров стенки тощей кишки. Кисты были иссечены, по результатам гистологического исследования, получена морфологическая картина лимфангиолейомиоматоза – образование было представлено щелевидными сосудами различной величины, стенка которых представлялась светлыми эпителиойдныи клетками. С учетом данных анамнеза пациентки: женщина 34 лет, не курит, с первичным бесплодием, рецидивирующим пневмотораксом, вентиляционными нарушениями и характерной КТ–картиной, – диагноз лимфангиолейомиоматоза не вызывал сомнений.
Диагноз больной: лимфангиолейомиоматоз с поражением легких, множественными лимфангиолейомиомами брюшной полости и забрюшинного пространства. Рецидивирующий пневмоторакс в анамнезе.
ЛАМ является редким мультисистемным заболеванием, поражающим преимущественно женщин в период пременопаузы и характеризующимся кистозной трансформацией легких, поражением лимфатической системы и новообразованиями в брюшной полости (ангиомиолипомами) [1–4]. Для ЛАМ характерна пролиферация патологических гладкомышечных клеток в стенках кровеносных и лимфатических капиллярах, интерстициальной ткани, приводящая к формированию кист в легких и кистозных структур, заполненных лимфатической жидкостью (лимфангиолейомиомы) по ходу осевого лимфатического протока.
Выделяют спорадический ЛАМ (без признаков генетического заболевания) и ЛАМ, ассоциированный с туберозным склерозом (ТС) [6–8] – наследственным аутосомно–доминантным заболеванием, характеризующимся умственной отсталостью, развитием опухолевидных образований (гамартом) в различных органах, встречающимся с частотой 1 на 5800 новорожденных [9]. ЛАМ встречается, по разным источникам, от 1 до 2,6 случаев на 1 млн женщин спорадически [2,10] и у каждой третьей пациентки с ТС [6–8]. Заболевание манифестирует чаще всего в детородном возрасте и периоде пременопаузы, в среднем в 34 года [11]. В литературе встречаются казуистические описания ЛАМ, ассоциированного с ТС у мужчин и детей, и нет ни одного описания спорадического такого случая [12].
Первый зарегистрированный случай ЛАМ (смерть женщины с ТС от двустороннего пневмоторакса) описан Lutembacher в 1918 г. [13]. Первое сообщение о спорадическом ЛАМ было в 1937–м Von Stossel [14], который описал молодую женщину, погибшую от острой дыхательной недостаточности с множественными кистами обоих легких и расширенным грудным лимфатическим протоком, автор назвал болезнь «мышечным циррозом». В 1966 г. Cornog и Enterline [15] рассмотрели 20 клинических случаев ЛАМ, сопровождавшегося образованиями брюшной полости, названными авторами лимфангиомиомами.
В 1975 г. Corrin и др. [16] предположили связь между ЛАМ и ТС. К 1990 г. в литературе уже имелись описания ЛАМ из различных стран: США [17,18], Великобритания [10], Франция [2], Юго–Восточная Азия [1]. В 2000 г. Carsillo и Astrinidis [19] выявили наличие при ЛАМ соматических мутаций в генах TSC1 и TSC2 ТС.
Выделяют 2 типа патологических клеток при ЛАМ: малые веретенообразные клетки, имеющие высокую пролиферативную активность, и большие эпителиоидноподобные клетки, богатые цитоплазмой. Оба вида этих клеток реагируют с антигладкомышечными антителами к a–актину, виментину, десмину [20]. Эпителиоидные клетки ЛАМ в своем составе имеют электронно плотные гранулы с тирозиназой и включениями связанного с меланомой человека гликопротеина 100 (НМВ–45) [11,21]. В клетках обоих типов при ЛАМ также обнаруживаются рецепторы к эстрогену, прогестерону и факторам роста [23–25 ]. Существуют сведения о возможной способности клеток ЛАМ к метастазированию: одинаковые мутации в TSC2 обнаруживаются у пациентов со спорадическим ЛАМ и в донорском легком после его трансплантации от здорового донора [26–27], а также в крови, моче, плевральных и асцитических хилезных выпотах больных ЛАМ [28]. Первоначальный источник клеток ЛАМ неизвестен, но потенциальными считаются ангиомиолипомы (хотя у 50% спорадического ЛАМ не удается обнаружить признаки ангиомиолипомы) и лимфатическая система [29].
Туберозный склероз (ТС) – это наследственное аутосомно–доминантное заболевание, обусловленное герминативными мутациями в одном из двух генов TSC1 и TSC2 [30,31]. По литературным данным, у 2/3 больных ТС является результатом спорадических мутаций в вышеуказанных генах и нет данных за отягощенный семейный анамнез [32]. Причиной развития спорадического ЛАМ являются соматические мутации в генах ТС – TSC1 и TSC2, наиболее часто встречающиеся в TSC2 [19, 33].
Ген TSC1 расположен на 9–й хромосоме (9q34) и содержит 23 экзона [34]. TSC1 кодирует белок–гамартин [35], участвующий в трансформации структуры белка актина [36]. Ген TSC2 расположен на 16–й хромосоме (16p13.3) и содержит 41 экзона [37], TSC2 кодирует белок туберин, играющий определенную негативную роль в клеточном цикле, ее росте и пролиферации [32,35]. Туберин интегрирует сигналы от факторов роста и источников энергии. Посредством последующего включения в патогенез фосфатидилинозитол–3–ОН–киназы (PI3K), сывороточной треонин–киназы (Akt), рапамицина (mTOR), их фосфорилирования, активации и/или ингибиции, преобразование белка Rheb–GTP в Rheb–GDP приводит в конечном счете к атипичной клеточной пролиферации и ангиогенезу [38,39].
Первыми клиническими проявлениями ЛАМ обычно являются одышка, внезапный пневмоторакс, хилезные плевральные выпоты и интраабдоминальные кровотечения [1–5]. Одышка, кашель и кровохарканье – наиболее частые легочные проявления ЛАМ [5]. Одышка встречается у примерно 70% пациентов с ЛАМ и является следствием кистозной трансформации легочной ткани [5]. Пневмоторакс, который может периодически рецидивировать, развивается приблизительно у 2/3 больных ЛАМ и существенно осложняет течение заболевания [4]. Основным клиническим проявлением ЛАМ при КТ является наличие хорошо отграниченных тонкостенных округлых или овальных кист, расположенных в обоих легких [40–42]. Эти кисты могут быть различного размера (от нескольких мм до нескольких см) и количества (от нескольких рассеянных кист до полного замещения нормальной паренхимы легких кистами). Большинство кист имеют круглую форму и тонкую, хорошо визуализирующуюся стенку. У существенного количества пациентов с ЛАМ можно выявить очаги бронхиолита, окруженного клетками ЛАМ [16]. Единственный самый важный диагностический тест – это КТ с высокой разрешающей способностью. Если при КТ выявляется характерная картина кистозного преобразования легких, в анамнезе имеет место пневмо– или хилоторакс, при исследовании функции внешнего дыхания выявляются обструктивные вентиляционные нарушения и снижение диффузионной способности легких нет никакой необходимости в проведении биопсии легких для подтверждения диагноза [11]. У больных ТС с выявленными при КТ кистами легких диагноз ЛАМ очень вероятен, также как и одновременное обнаружение ангиомиолипомы и кистозной трансформации легких. При обнаружении кист в легких необходимо проводить дифференциальный диагноз с другими редкими заболеваниями с подобной симптоматикой – синдромом Birt–Hogg–Dube [43–44], Sjogren [45–46], гистиоцитозом Х [47]. При этом в сложных клинических случаях рекомендуется даже проведение открытой биопсии легких совместно с видеторакоскопией и иммунофлюоресцентное окрашивание полученного материала на a–актин, десмин, виментин и иммуногистохимическое исследование на наличие антител НМВ–45 [21,48,49].
К экстрапульмональными проявлениям ЛАМ относятся: лимфоаденопатия, объемные кистозные лимфатические массы (лимфангиолейомиомы), асцит [50,51] и доброкачественные опухоли (ангиомиолипомы). Ангиомиолипома – это доброкачественная опухоль, имеющая смешанное мезенхимальное происхождение, обычно локализующаяся в почках и встречающаяся примерно в у 80% пациентов с ЛАМ, ассоциированным с ТС, и в 40% случаях спорадического ЛАМ [5,51]. Размеры этих опухолей колеблются от 1 мм до 20 см в диаметре [52,53], большинство бессимптомны [51,54,55]. При КТ эти образования дают характерную картину – неоднородной плотности с участками нормальной почечной паренхимы и жировой ткани [51,52], хорошо васкуляризированы. Хотя часто ангиомиолипомы встречаются в отсутствие ЛАМ, но их обнаружение при КТ, направляет специалиста на более детальное исследование легких на предмет наличия кист.
Увеличенные лимфоузлы брюшной полости, забрюшинного пространства и таза также хорошо визуализируются при КТ у 1/3 больных ЛАМ и редко себя проявляют клинически [51]. Лимфангиолейомиомы – это большие кистозные массы, встречающиеся в брюшной полости, забрюшинном пространстве, тазу, средостении и шее, проявляющиеся тошнотой, чувством распирания в брюшной полости, болями различной локализации и дизурическими расстройствами, иногда усиливающимися к концу дня. Это связано с увеличением в размерах абдоминальных лимфангиолейомиом из–за накопления лимфы в нижних конечностях к концу дня [56]. У 10% больных ЛАМ развивается асцит в связи с обструкцией лимфатических сосудов [4,51]. Отмечено увеличение распространенности менингиом у пациентов в ЛАМ по сравнению с общей популяцией [57]. Связано ли это с ЛАМ, ТС, лечением прогестероном, неизвестно.
В среднем от момента первых клинических проявлений до точного установления диагноза ЛАМ проходит 5–6 лет [58]. У около 50% пациентов с ЛАМ имело место развитие пневмоторакса [59], хилоторакса с или без асцита [60,61], случайного обнаружения торакоабдоминальной лимфоаденопатии и лимфангиолейомиом [50,60], предположение о наличии лимфопролиферативного заболевания [62,63]. В редких случаях ЛАМ также может манифестировать с клиники абдоминальной боли различной интенсивности вплоть до развития «острого живота» [64]. Диагноз ЛАМ следует предполагать у молодой женщины с клиникой пневмоторакса (или в анамнезе) и постоянной одышкой или кровохарканьем и у молодой некурящей женщины с эмфиземой легких с обструктивными вентиляционными нарушениями и поражением ткани легких [11].
Данные объективного осмотра пациентов с ЛАМ могут оставаться нормальными. При прогрессировании заболевания выявляется плевральный выпот, образования брюшной полости (ангиомиолипомы, лимфангиолейомиомы), при ЛАМ, ассоциированном с ТС – умственная отсталость, подногтевые фибромы, нейрофибромы, ангиофибромы лица, шагреневые пятна на коже.
При ЛАМ чаще всего выявляют вентиляционные нарушения по обструктивному типу и снижение диффузионной способности легких. Ограничение воздушного потока выявляется у более чем 60% пациентов. Снижение диффузионной способности легких встречается у таких пациентов так же часто, как и снижение ОФВ1 [5]. Вентиляционные нарушения по рестриктивному типу обычно развиваются у пациентов с плевральными выпотами в анамнезе или перенесшими плевродез [65].
В большинстве случаев ЛАМ – это хроническое заболевание, охватывающее десятилетия. Даже тяжелое течение ЛАМ может стабилизироваться и не проявлять в последующем никаких респираторных нарушений. Ретроспективный анализ 402 пациентов с ЛАМ, начатый с 1995 г. в Великобритании, показал, что к 2006 г. умерло 22 человека, 8 из которых перенесли трансплантацию легких. Летальность в этом контингенте составила 5,5%. Из 380 выживших пациентов 38 (10%) перенесли трансплантацию легких, а 85% пациентов выжили и без трансплантации. По данным литературы, в США десятилетняя выживаемость пациентов с ЛАМ составляет более 90% [3]. По результатам других исследований, 10–летняя выживаемость составляет 55–71% [2,18]. Регулярные инструментальные исследования помогают контролировать прогрессирование заболевания. В частности, изменения ОВФ1 и DLCO коррелируются с признаками прогрессированием заболевания, выявленными при КТ, путем определения LHS и другими методами [65]. LHS (LAM histology score) – это объем пораженной кистами легочной ткани, определяемый во время открытой биопсии легких [66]. Выделяют: LHS–1 ≤25%, LHS–2 = 25–50% и LHS–3 ≥50%. Определение LHS имеет важное прогностическое значение, поскольку имеются различные данные по выживаемости пациентов в зависимости от значения этого показателя [66].
Анализ наблюдения 275 пациентов в течение 4 лет [58] показал средние ежегодные показатели снижения ОФВ1 и DLCO – 75±9 мл (1,7±0,4%) и 0,69±0,07 мл/мин/мм рт.ст. (2,4±0,4%) соответственно. Ежегодные нормы снижения этих показателей зависят от возраста, факта курения, сопутствующей патологии [67,68]. Например, ежегодная норма снижения DLCO для некурящих женщин составляет 0,28–0,29 мл/мин/мм рт.ст. [69]. Для сравнения, средние нормы снижения ОФВ1 и DLCO для пациентов с a1–антитрипсиновой недостаточностью составляют соответственно 67±14 мл/год и 1,07±0,21 мл/мин/мм рт.ст. [70].
В настоящее время не существует эффективных методов лечения ЛАМ, которые бы полностью останавливали повреждение легких или вызывали обратное развитие процесса. Поскольку ЛАМ – это заболевание женщин преимущественно в предклимактерическом периоде, прогрессирующее во время беременности [71] или после терапии эстрогенами [72], правомерно предполагать эффективность гормональной терапии при ЛАМ. Проводилось много исследований по изучению эффективности различной заместительной терапии. Существуют несколько сообщений об уменьшении объема плевральных выпотов после двусторонней овариоэктомии [73], но в исследованиях Taylor и др. у 15 пациентов после овариоэктомии не было получено таких результатов [18]. В настоящее время не существует объективных признаков эффективности антиэстрогеновой терапии [73]. В литературе существуют сообщения об эффективности применения аналогов гонадотропин–релизинг гормона [73], но в других исследованиях не было получено таких убедительных результатов [74]. В ретроспективном исследовании Johnson и Tattersfield [67] пациентки, принимавшие прогестерон, показывали более медленное снижение показателей ОФВ1 и DLCO по сравнению с нелеченными женщинами. Однако в другом исследовании 275 пациентов с ЛАМ [58] не было отмечено существенной разницы скорости ежегодного снижения показателей ОФВ1 и DLCO между пациентками, получающими прогестерон и находящимися без лечения. Имеются также сведения о более быстром ежегодном снижении ОФВ1 и DLCO у пациентов получавших эстрогены [75,76]. Исследование влияния тамоксифена на течение ЛАМ также дало спорные результаты [58]. Однозначно ответить на вопрос об использовании гормональной терапии в качестве лечения ЛАМ в настоящее время не представляется возможным.
Приблизительно у 20% больных ЛАМ в процессе обследования выявляется положительная проба с бронхолитиками [65], у некоторых пациентов имеют место в анамнезе эпизоды дистантных хрипов. Рекомендуется к лечению таких пациентов добавлять бронхолитики. Однако имеются исследовательские работы, сообщающие о более быстром ежегодном снижении показателей ОФВ1 и DLCO у пациентов, получающих бронхолитическую терапию, по сравнению с пациентами без нее [65]. Причины такого результата остаются неизвестными. В литературе также нет данных о пользе ингаляционных или системных глюкокортикостероидов.
Достоверно эффективных методов лечения в настоящее время не существует, но ведется множество исследований с целью ответить на этот вопрос. Чаще всего лечение сводится к симптоматическому развившихся осложнений (пневмоторакс, хилезный плевральный выпот и экстрапульмональные проявления).
Пневмоторакс при ЛАМ имеет тенденцию к рецидивированию. Простая закрытая торакостомия может быть первоначальным методом лечения пневмоторакса, а при его рецидивировании необходимо проводить химический или хирургический плевродез [59]. Плевродез по Talc является наиболее эффективным методом химического плевродеза, но часто приводящего к формированию фиброторакса, осложняющего отделение легкого при его трансплантации и способствующего развитию более серьезных послеоперационных осложнений [59].
Другие проявления ЛАМ – асцит и хилоторакс – также являются трудной терапевтической задачей. С учетом механизмов образования хилоторакса при ЛАМ (обструкция грудного протока, трансдиафрагмальное пропотевание хилезной асцитической жидкости), лечение его сводится к плевродезу, плевроцентезу и перевязке грудного протока [11]. Лечение хилоторакса, не приводящего к развитию одышки, не требуется [11]. Повторные плевроцентезы могут способствовать развитию инфекционных осложнений. Пациентам с хилотораксом рекомендуется соблюдать диету с ограничением потребления растительных и животных жиров [61]. При первичном развитии хилоторакса рекомендуется проведение плевроцентеза, а при рецидивирующем – хирургический или химический плевродезы. До, во время и после проведения плевродеза пациента необходимо перевести на парентеральное питание, что впоследствии будет способствовать более равномерному сращению париетальной и висцеральной плевры, препятствовать образованию плевральных карманов [61]. В литературе также рассматриваются другие методы лечения хилоторакса и асцита при ЛАМ: медроксипрогестероном [77], формированием плевроперитонеальных шунтов [78], но на практике эти методы редко используются. Сдавление постепенно увеличивающимися в размерах лимфангиолейомиомами мочевого пузыря, кишечника, тазовых вен способствуют появлению боли, стойкого запора, дизурических расстройств и периферических отеков [79]. В литературе имеются данные об успешном применении в таких ситуациях соматостатина и октреотида [80], способствующих сокращению объема хилезных выпотов, хилурии, асцита и периферических отеков. У пациентов с ЛАМ необходимо определять сатурацию кислорода периферической крови. Кислородотерапия предотвращает развитие гипоксемии и препятствует прогрессированию легочной гипертензии.
Пациентам с тяжелым ЛАМ, находящимся на постоянной кислородотерапии, и со снижением DLCO и/или ОФВ1 на 40% и более от должных значений рекомендуется рассмотреть вопрос о трансплантации легких с учетом возможных послеоперационных осложнений вплоть до летального исхода [81,82]. Имеются сообщения о рецидиве ЛАМ в пересаженном легком, что происходит из–за «метастатического» распространения оставшихся клеток ЛАМ [83,84].
Очень часто ангиомиолипомы почек диагностируют как злокачественные опухоли почек с последующей нефрэктомией. Часто даже у пациентов с двусторонней локализацией ангиомиолипом сохраняется нормальное функционирование почек. Основные показания для хирургического лечения ангиомиолипом – кровоточащие образования диаметром более 4 см в диаметре, приводящие к анемизации. Типичными клиническими проявлениями в этой ситуации являются боль в поясничной области с или без наличия гематурии. Хирургическое лечение ангиомиолипом направлено на максимальное сохранении функций почек и включает резекцию и локальную эмболизацию опухолевых образований [52]. Пациенты с АМЛ почек без признаков кровотечения должны наблюдаться у уролога с возможным применением рентгенологических методов лечения. АМЛ необходимо дифференцировать со злокачественными новообразованиями почек. При наличии признаков быстрого роста образования, неточных данных о его структуре и типе роста рекомендуется проведение биопсии и КТ контроль каждые 6 мес. [52]. ЛАМ – это мультисистемное заболевание, поражающее преимущественно женщин, характеризующееся кистозным преобразованием легких (приводящим к острой дыхательной недостаточности), ангиомиолипомами (приводящими к внутрибрюшным кровотечениям) и поражением лимфатической системы (с развитием асцита и хилоторакса). В настоящее время нет достоверно эффективных методов лечения. Но прогресс в цитобиологии, особенно в изучении механизмов клеточной пролиферации и роста и факторов, регулирующих лимфо– и ангиогенез, дает надежды на открытие эффективных методов лечения ЛАМ. Основным направлением этих исследований является изучение схем запрета mTOR, матрицы металлопротеиназы (ММР) и ангиогенеза. Поскольку mTOR является пусковым моментом в патогенезе ЛАМ, следовательно его антагонисты могут быть достаточно эффективны. Рапамицин – это антибактериальный препарат, обладающий также и противогрибковой активностью и относящийся к группе макролидов, зарегистрированный в США. По результатам уже проведенных исследований, на фоне двухмесячной терапии рапамицином происходит значительное уменьшение в размерах опухолей почки у лабораторных крыс [84,85]. В настоящее время в ходу исследования Cincinnati Angiomyolipoma Sirolimus (CAST) и Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES), изучающие влияния рапамицина на ангиомиолипомы почек и легкие соответственно.
ММР играет важную роль в процессе лимфангиогенеза [86,87]. Поражение легочной ткани при ЛАМ происходит в результате пролиферации гладкомышечных клеток ЛАМ и под влиянием токсических веществ, ими выделяемых [88–89]. В легких клетки ЛАМ выявляются в стенках кист и интерстиции. Предполагают, что чрезмерная продукция матриксных металлопротеиназ (ММР) клетками ЛАМ может способствовать разрушению интерстициальной ткани легкого, приводящему к его деструкции [87–89]. С ЛАМ связывают несколько ММР: ММР–2 (желатиназа А), ММР–1, ММР–9 (желатиназа В) и ММР–14 [88,89]. Антагонистом активности ММР является доксициклин, препятствующий росту и миграции опухолевых клеток, лимф– и ангиогенезу и пролиферации гладкомышечных клеток [90–92]. Таким образом, ингибиторы матриксных металлопротеиназ могут быть основой в лечении опухолевых заболеваний [90,91]. Moses и др. опубликовали результаты лечения пациента с тяжелым ЛАМ в течение 3 мес. доксициклином, после чего значительно снизилось содержание ММР с одновременным улучшением показателей спирографии и сатурации крови [92–93].
Авиаперелеты и беременность несколько увеличивает риск развития осложнений ЛАМ (пневмо– и хилоторакс, рост ангиомиолипомы почек) [Ч 94]. У некоторых пациенток дебют заболевания также был связан с беременностью [2,6]. Однако у большого количества женщин с ЛАМ беременность протекала без осложнений. Риск развития рецидива пневмоторакса при авиаперелетах увеличивается у пациентов с уже перенесенным пневмотораксом в анамнезе и уменьшается после проведенного плевродеза [11]. Авиаперелеты запрещены больным с пневмотораксом, а при появлении новых симптомов для его исключения необходимо перед полетом провести рентгенографию легких.
Таким образом, многие аспекты ЛАМ до сих пор остаются неясными. Тщательные клинические и молекулярные исследования в этой области необходимы для выявления эффективных методов лечения.


Литература
1. Kitaichi M, Nishimura K, Itoh H, et al. Pulmonary lymphangiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med. 1995;151:527–533.
2. Urban T, Lazor R, Lacronique J, et al. Pulmonary lymphangioleiomyomatosis: a study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM“O”P). Medicine (Baltimore). 1999;78:321–337.
3. Chu SC, Horiba K, Usuki J, et al. Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest. 1999;115:1041–1052.
4. Johnson SR, Tattersfield AE. Clinical experience of lymphangioleiomyomatosis in the UK. Thorax. 2000;55:1052–1057.
5. Ryu JH, Moss J, Beck GJ, et al. The NHLBI Lymphangioleiomyomatosis Registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med. 2006;173:105–111. Epub 2005 Oct 6.
6. Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex. Mayo Clin Proc. 2000;75:591–594.
7. Moss J, Avila NA, Barnes PM, et al. Prevalence and clinical characteristics of lymphangioleiomyomatosis (LAM) in patients with tuberous sclerosis complex. Am J Respir Crit Care Med. 2001;164:669–671.
8. Franz DN, Brody A, Meyer C, et al. Mutational and radiographic analysis of pulmonary disease consistent with lymphangioleiomyomatosis and micronodular pneumocyte hyperplasia in women with tuberous sclerosis. Am J Respir Crit Care Med. 2001;164:661–668.
9. Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann NY Acad Sci. 1991;615:125–127.
10. Johnson SR, Tattersfield AE. Decline in lung function in lymphangioleiomyomatosis: relation to menopause and progesterone treatment. Am J Respir Crit Care Med 1999; 160: 628–633.
11. Johnson SR. Lymphangioleiomyomatosis. Am J Respir Crit Care Med 2006; 27: 1056–1065.
12. Aubry MC, Myers JL, Ryu JH, et al. Pulmonary lymphangioleiomyomatosis in a man. Am J Respir Crit Care Med 2000; 162: 749–752.
13. Lutembacher R. Dysembryomes metatypiques des reins. Carcinose sublimiare aigue du poumon avec emphyseme generalise et double pneumothorax [Atypical kidney dysembryoma. Acute submiliary lung carcinomatosis with generalised emphysema and double pneumothorax]. Ann Med 1918; 5: 435.
14. von Stossel E. Uber muskulare Cirrhose der Lunge [Muscular cirrhosis of the lungs]. Beitr Klin Tuberk 1937; 90: 432–442.
15. Cornog JL Jr, Enterline HT. Lymphangiomyoma, a benign lesion of chyliferous lymphatics synonymous with lymphangiopericytoma. Cancer 1966; 19: 1909–1930.
16. Corrin B, Liebow AA, Friedman PJ. Pulmonary lymphangioleiomyomatosis. Am J Pathol 1975; 79: 348–382.
17. Chu SC, Horiba K, Usuki J, et al. Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest 1999; 115: 1041–1052.
18. Taylor JR, Ryu J, Colby TV, Raffin TA. Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J Med 1990; 323: 1254–1260.
19. Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA 2000; 97: 6085–6090.
20. Ferrans VJ, Yu ZX, Nelson WK, et al. Lymphangioleiomyomatosis (LAM): a review of clinical and morphological features. J Nippon Med Sch. 2000;67:311–329.
21. Matsumoto Y, Horiba K, Usuki J, Chu SC, Ferrans VJ, Moss J. Markers of cell proliferation and expression of melanosomal antigen in lymphangioleiomyomatosis. Am J Respir Cell Mol Biol 1999; 21: 327–336.
22. Ohori NP, Yousem SA, Sonmez–Alpan E, et al. Estrogen and progesterone receptors in lymphangioleiomyomatosis, epithelioid hemangioendothelioma, and sclerosing hemangioma of the lung. Am J Clin Pathol. 1991;96:529–535.
23. Valencia JC, Matsui K, Bondy C, et al. Distribution and mRNA expression of insulin–like growth factor system in pulmonary lymphangioleiomyomatosis. J Investig Med. 2001;49:421–433.
24. Matsui K, Takeda K, Yu ZX, et al. Downregulation of estrogen and progesterone receptors in the abnormal smooth muscle cells in pulmonary lymphangioleiomyomatosis following therapy. An immunohistochemical study. Am J Respir Crit Care Med 2000; 161: 1002–1009.
25. Banner AS. Hormone receptors in lymphangioleiomyomatosis. Chest 1984; 85: 3–4.
26. Bittmann I, Rolf B, Amann G, et al. Recurrence of lymphangioleiomyomatosis after single lung transplantation: new insights into pathogenesis. Hum Pathol. 2003;34:95–98.
27. Karbowniczek M, Astrinidis A, Balsara BR, et al. Recurrent lymphangioleiomyomatosis after transplantation: genetic analyses reveal a metastatic mechanism. Am J Respir Crit Care Med. 2003;167:976–982. Epub 2002 Oct 31.
28. Crooks DM, Pacheco–Rodriguez G, DeCastro RM, et al. Molecular and genetic analysis of disseminated neoplastic cells in lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2004:101:17462–17467. Epub 2004 Dec 6.
29. Henske EP. Metastasis of benign tumor cells in tuberous sclerosis complex. Genes Chromosomes Cancer. 2003;38:376–381.
30. Sepp T, Yates JR, Green AJ. Loss of heterozygosity in tuberous sclerosis hamartomas. J Med Genet 1996; 33: 962–964.
31. Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 2001; 68: 64–80.
32. Roach ES, DiMario FJ, Kandt RS, Northrup H. Tuberous Sclerosis Consensus Conference: recommendations for diagnostic evaluation. National Tuberous Sclerosis Association. J Child Neurol 1999; 14: 401–407.
33. Astrinidis A, Khare L, Carsillo T, et al. Mutational analysis of the tuberous sclerosis gene TSC2 in patients with pulmonary lymphangioleiomyomatosis. J Med Genet 2000; 37: 55–57.
34. Maziak DE, Kesten S, Rappaport DC, Maurer J. Extrathoracic angiomyolipomas in lymphangioleiomyomatosis. Eur Respir J 1996; 9: 402–405.
35. Avila NA, Bechtle J, Dwyer AJ, Ferrans VJ, Moss J. Lymphangioleiomyomatosis: CT of diurnal variation of lymphangioleiomyomas. Radiology 2001; 221: 415–421.
36. Lemaitre L, Robert Y, Dubrulle F, et al. Renal angiomyolipoma: growth followed up with CT and/or US. Radiology 1995; 197: 598–602.
37. Steiner MS, Goldman SM, Fishman EK, Marshall FF. The natural history of renal angiomyolipoma. J Urol 1993; 150: 1782–1786
38. Boehler A, Speich R, Russi EW, Weder W. Lung transplantation for lymphangioleiomyomatosis. N Engl J Med 1996; 335: 1275–1280.
39. Berger JL, Shaff MI. Pulmonary lymphangioleiomyomatosis. J Comput Assist Tomogr 1981; 5: 565–567.
40. Merchant RN, Pearson MG, Rankin RN, et al. Computerized tomography in the diagnosis of lymphangioleiomyomatosis. Am Rev Respir Dis. 1985;131:295–297.
41. Avila NA, Chen CC, Chu SC, et al. Pulmonary lymphangioleiomyomatosis: correlation of ventilation–perfusion scintigraphy, chest radiography, and CT with pulmonary function tests. Radiology. 2000;214:441–446.
42. Avila NA, Kelly JA, Dwyer AJ, et al. Lymphangioleiomyomatosis: correlation of qualitative and quantitative thin–section CT with pulmonary function tests and assessment of dependence on pleurodesis. Radiology. 2002;223:189–197.
43. Butnor KJ, Guinee DG Jr. Pleuropulmonary pathology of Birt–Hogg–Dube syndrome. Am J Surg Pathol. 2006;30:395–399.
44. Painter JN, Tapanainen H, Somer M, et al. A 4–bp deletion in the Birt–Hogg–Dube gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet. 2005;76:522–527. Epub 2005 Jan 18
45. Koyama M, Johkoh T, Honda O, et al. Pulmonary involvement in primary Sjogren’s syndrome: spectrum of pulmonary abnormalities and computed tomography findings in 60 patients. J Thorac Imaging. 2001;16:290–296.
46. Lohrmann C, Uhl M, Warnatz K, et al. High–resolution CT imaging of the lung for patients with primary Sjogren’s syndrome. Eur J Radiol. 2004; 52:137–143.
47. Tazi A. Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J. 2006;27:1272–1285
48. Tanaka H, Imada A, Morikawa T, et al. Diagnosis of pulmonary lymphangioleiomyomatosis by HMB45 in surgically treated spontaneous pneumothorax. Eur Respir J 1995; 8: 1879–1882.
49. Bonetti F, Chiodera PL, Pea M, et al. Transbronchial biopsy in lymphangiomyomatosis of the lung. HMB45 for diagnosis. Am J Surg Pathol 1993; 17: 1092–1102.
50. Matsui K, Tatsuguchi A, Valencia J, et al. Extrapulmonary lymphangioleiomyomatosis (LAM): clinicopathologic features in 22 cases. Hum Pathol 2000; 31: 1242–1248.
51. Avila NA, Kelly JA, Chu SC, Dwyer AJ, Moss J. Lymphangioleiomyomatosis: abdominopelvic CT and US findings. Radiology 2000; 216: 147–153.
52. Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int. 2004;66:924–934
53. Jaiswal VR, Baird J, Fleming J, et al. Localized retroperitoneal lymphangioleiomyomatosis mimicking malignancy. A case report and review of the literature. Arch Pathol Lab Med. 2003;127:879–882.
54. Bernstein SM, Newell JD Jr, Adamczyk D, Mortenson RL, King TE Jr, Lynch DA. How common are renal angiomyolipomas in patients with pulmonary lymphangiomyomatosis? Am J Respir Crit Care Med 1995; 152: 2138–2143.
55. Maziak DE, Kesten S, Rappaport DC, Maurer J. Extrathoracic angiomyolipomas in lymphangioleiomyomatosis. Eur Respir J 1996; 9: 402–405.
56. Avila NA, Bechtle J, Dwyer AJ, Ferrans VJ, Moss J. Lymphangioleiomyomatosis: CT of diurnal variation of lymphangioleiomyomas. Radiology 2001; 221: 415–421.
57. Moss J, DeCastro R, Patronas NJ, et al. Meningiomas in lymphangioleiomyomatosis. JAMA. 2001;286:1879–1881.
58. Taveira–Dasilva AM, Stylianou MP, Hedin CJ, et al. Decline in lung function in patients with lymphangioleiomyomatosis treated with or without progesterone. Chest. 2004;126:1867–1874.
59. Almoosa KF, Ryu JH, Mendez J, et al. Management of pneumothorax in lymphangioleiomyomatosis: effects on recurrence and lung transplantation complications. Chest. 2006;129:1274–1281.
60. Matsui K, Tatsuguchi A, Valencia J, et al. Extrapulmonary lymphangioleiomyomatosis (LAM): clinicopathologic features in 22 cases. Hum Pathol. 2000;31:1242–1248.
61. Ryu JH, Doerr CH, Fisher SD, et al. Chylothorax in lymphangioleiomyomatosis. Chest. 2003;123:623–627.
62. Jaiswal VR, Baird J, Fleming J, et al. Localized retroperitoneal lymphangioleiomyomatosis mimicking malignancy. A case report and review of the literature. Arch Pathol Lab Med. 2003;127:879–882.
63. Llopis I, Arandiga R, Real E, et al. Lymphangiomyomatosis mimicking an abdominal lymphoma. Haematologica. 2002;87:EIM23.
64. Lu HC, Wang J, Tsang YM, et al. Lymphangioleiomyomatosis initially presenting with abdominal pain: a case report. Clin Imaging. 2003;27: 166–170.
65. Taveira–DaSilva AM, Hedin C, Stylianou MP, et al. Reversible airflow obstruction, proliferation of abnormal smooth muscle cells, and impairment of gas exchange as predictors of outcome in lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2001;164:1072–1076.
66. Matsui K, Beasley MB, Nelson WK, et al. Prognostic significance of pulmonary lymphangioleiomyomatosis histologic score. Am J Surg Pathol. 2001;25:479–484.
67. Johnson SR, Tattersfield AE. Decline in lung function in lymphangioleiomyomatosis: relation to menopause and progesterone treatment. Am J Respir Crit Care Med. 1999;160:628–633.
68. Burrows B, Lebowitz MD, Camilli AE, et al. Longitudinal changes in forced expiratory volume in one second in adults: methodologic considerations and findings in healthy nonsmokers. Am Rev Respir Dis. 1986;133: 974–980.
69. Sherrill DL, Enright PL, Kaltenborn WT, et al. Predictors of longitudinal change in diffusing capacity over 8 years. Am J Respir Crit Care Med. 1999;160:1883–1887.
70. Dowson LJ, Guest PJ, Stockley RA. Longitudinal changes in physiological, radiological, and health status measurements in alpha(1)–antitrypsin deficiency and factors associated with decline. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1805–1809.
71. 63. Brunelli A, Catalini G, Fianchini A. Pregnancy exacerbating unsuspected mediastinal lymphangioleiomyomatosis and chylothorax. Int J Gynaecol Obstet. 1996;52:289–290.
72. Yano S. Exacerbation of pulmonary lymphangioleiomyomatosis by exogenous oestrogen used for infertility treatment. Thorax. 2002;57:1085– 1086.
73. de la Fuente J, Paramo C, Roman F, et al. Lymphangioleiomyomatosis: unsuccessful treatment with luteinizing–hormone–releasing hormone analogues. Eur J Med. 1993;2:377–378.
74. Shen A, Iseman MD, Waldron JA, King TE. Exacerbation of pulmonary lymphangioleiomyomatosis by exogenous estrogens. Chest 1987; 91: 782–785.
75. Yano S. Exacerbation of pulmonary lymphangioleiomyomatosis by exogenous oestrogen used for infertility treatment. Thorax 2002; 57: 1085–1086.
76. Druelle S, Aubry P, Levi–Valensi P. Pulmonary lymphangiomyomatosis: a 3–year follow–up of medroxyprogesterone acetate therapy. Apropos of a case [in French]. Rev Pneumol Clin. 1995;51:284–287.
77. Boudard C, Gamondes JP, Mornex JF, et al. Pleuro–peritoneal Denver shunt in treatment of chronic pleurisy [in French]. Ann Chir. 1998;52: 192–196.
78. Kimura M, Morikawa T, Takeuchi K, et al. Lymphangioleiomyomatosis with chylous ascites treatment successfully by peritoneo–venous shunting [in Japanese]. Nihon Kyobu Shikkan Gakkai Zasshi. 1996;34:557–562
79. Makrilakis K, Pavlatos S, Giannikopoulos G, et al. Successful octreotide treatment of chylous pleural effusion and lymphedema in the yellow nail syndrome. Ann Intern Med. 2004;141:246–247.
80. Pechet TT, Meyers BF, Guthrie TJ, et al. Lung transplantation for lymphangioleiomyomatosis. J Heart Lung Transplant. 2004;23:301–308.
81. Kpodonu J, Massad MG, Chaer RA, et al. The US experience with lung transplantation for pulmonary lymphangioleiomyomatosis. J Heart Lung Transplant. 2005;24:1247–1253.
82. Nine JS, Yousem SA, Paradis IL, Keenan R, Griffith BP. Lymphangioleiomyomatosis: recurrence after lung transplantation. J Heart Lung Transplant 1994; 13: 714–719.
83. Karbowniczek M, Astrinidis A, Balsara BR, et al. Recurrent lymphangiomyomatosis after transplantation: genetic analyses reveal a metastatic mechanism. Am J Respir Crit Care Med 2003; 167: 976–982.
84. Kenerson H, Dundon TA, Yeung RS. Effects of rapamycin in the Eker rat model of tuberous sclerosis complex. Pediatr Res. 2005;57:67–75. Epub 2004 Nov19.
85. Yeung RS. Multiple roles of the tuberous sclerosis complex genes. Genes Chromosomes Cancer. 2003;38:368–375.
86. Shapiro SD. Chairman’s summary. Proc Am Thorac Soc. 2006;3: 397–400.
87. Ji RC. Lymphatic endothelial cells, lymphangiogenesis, and extracellular matrix. Lymphat Res Biol. 2006;4:83–100.
88. Hayashi T, Fleming MV, Stetler–Stevenson WG, et al. Immunohistochemical study of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) in pulmonary lymphangioleiomyomatosis (LAM). Hum Pathol. 1997;28:1071–1078.
89. Matsui K, Takeda K, Yu ZX, et al. Role for activation of matrix metalloproteinases in the pathogenesis of pulmonary lymphangioleiomyomatosis. Arch Pathol Lab Med. 2000;124:267–275.
90. Duivenvoorden WCM, Popovic SV, Lhotak S, et al. Doxycycline decreases tumor burden in a bone metastasis model of human breast cancer. Cancer Res. 2002;62:1588–1591.
91. Chhipa RR, Singh S, Surve SV, et al. Doxycycline potentiates antitumor effect of cyclophosphamide in mice. Toxicol Appl Pharmacol. 2005; 202:268–277.
92. Franco C, Ho B, Mulholland D, et al. Doxycycline alters vascular smooth muscle cell adhesion, migration, and reorganization of fibrillar collagen matrices. Am J Pathol. 2006;168:1697–1709.
93. Moses MA, Harper J, Folkman J. Doxycycline treatment for lymphangioleiomyomatosis with urinary monitoring for MMPs. N Engl J Med. 2006;354:2621–2622.
94. McLoughlin L, Thomas G, Hasan K. Pregnancy and lymphangioleiomyomatosis: anaesthetic management. Int J Obstet Anesth 2003; 12: 40–44.



