




Введение
В 1907 г. американский патологоанатом Джордж Х. Уиппл (George Hoit Whipple), работавший в госпитале Дж. Гопкинса, опубликовал секционное наблюдение ранее неизвестного заболевания у 36–летнего мужчины. В течение 5 лет больной, врач по профессии, страдал лихорадкой, артритом, стойким и упорным кашлем, диареей и прогрессирующей потерей веса. При вскрытии обнаружено выраженное увеличение мезентеральных лимфатических узлов в сочетании с полисерозитом. При гистологическом исследовании кишечника и лимфоузлов выявлены множественные отложения липидов и большое число макрофагов, содержащих аргирофильные палочкообразные структуры. В качестве наиболее вероятной причины болезни Уиппл указал на нарушение метаболизма липидов и предложил назвать заболевание интестинальной липодистрофией. В то же время он не исключал и инфекционную этиологию заболевания [1].
В 1907 г. американский патологоанатом Джордж Х. Уиппл (George Hoit Whipple), работавший в госпитале Дж. Гопкинса, опубликовал секционное наблюдение ранее неизвестного заболевания у 36–летнего мужчины. В течение 5 лет больной, врач по профессии, страдал лихорадкой, артритом, стойким и упорным кашлем, диареей и прогрессирующей потерей веса. При вскрытии обнаружено выраженное увеличение мезентеральных лимфатических узлов в сочетании с полисерозитом. При гистологическом исследовании кишечника и лимфоузлов выявлены множественные отложения липидов и большое число макрофагов, содержащих аргирофильные палочкообразные структуры. В качестве наиболее вероятной причины болезни Уиппл указал на нарушение метаболизма липидов и предложил назвать заболевание интестинальной липодистрофией. В то же время он не исключал и инфекционную этиологию заболевания [1].Следует отметить, что впоследствии Дж. Уиппл опубликовал 270 работ, но больше к данному заболеванию не обращался. В 1934 г. он стал первым в США лауреатом Нобелевской премии в области медицины за разработку проблем лечения и профилактики пернициозной анемии. Несмотря на это, до сих пор его имя тесно ассоциируется с описанным им заболеванием – болезнью Уиппла (БУ).
Этиология и патогенез
В своем оригинальном описании случая Уиппл указывал на сходство палочковидных микробов, найденных им в собственной пластинке 12–перстной кишки, с бледной спирохетой. В то время инфекционная этиология болезни упорно оспаривалась оппонентами по причине ее несоответствия постулатам Коха. Однако, начиная с середины ХХ века, постепенно накапливались факты, свидетельствующие в пользу бактериальной этиологии заболевания. В 1949 г. было установлено, что в биоптатах лимфоузлов и тонкого кишечника содержатся PAS– (periodic acid Schiff) – позитивные макрофаги с включениями, похожими на продукты распада бактерий [2]. Спустя три года был описан первый случай успешного применения антибиотиков у больного БУ с быстрым обратным развитием клинической симптоматики и патоморфологических признаков. В начале 60–х гг. описаны палочкообразные тельца, выявленные в цитоплазме макрофагов при электронной микроскопии [3]. И только в работах последнего десятилетия [4–6] с помощью полимеразной цепной реакции (ПЦР) из инфицированных тканей пациентов была выделена грамположительная бацилла, получившая название Tropheryma whippelii (от греческого trophe – питание, eryma – барьер). По своим филогенетическим данным T. whippelii принадлежит к актиномицетам, имеет размеры 0,2х2,0 mм, трехслойную клеточную стенку, единственную круговую хромосому и небольшой размер генома (925 кД). Полагают, что патогенными могут быть не все, а строго определенные штаммы возбудителя.
В патогенезе заболевания наряду с генерализацией инфекции большое значение придается иммунологическим нарушениям. В пользу этого свидетельствуют наблюдаемые у таких больных снижение Т– клеточного соотношения СD4+/CD8+ и экспрессии CD11, уменьшение продукции интерлейкина–12 и g–интерферона. Параметры гуморального иммунитета изменяются мало. Гипотетический иммунологический дефект выражен достаточно слабо и весьма специфичен по отношению к T. whippelii, поскольку у пациентов с БУ частота возникновения других инфекций не превышает таковую в популяции. Показано, что T. whippelii накапливаются и размножаются в макрофагах. Таким образом, макрофаги, несмотря на сохранную функцию фагоцитоза, теряют способность к лизису возбудителя [7].
Эпидемиология
БУ относится к редким заболеваниям. К 2002 г. в литературе имелось чуть более 2000 наблюдений. Наиболее часто БУ выявляется у представителей белой расы – жителей Центральной Европы (55%) и Северной Америки (38%) . Единичные случаи болезни описаны среди испанцев, негров, индусов и представителей монголоидной расы. Преобладают жители сельской местности, чаще – фермеры. Мужчины поражаются в 8 раз чаще женщин, средний возраст к началу заболевания составляет 50 лет. Имеются отдельные сообщения о семейных случаях БУ. На возможную генетическую компоненту в развитии заболевания указывает повышенная частота обнаружения HLA–B27 (26–40%) у этих пациентов, однако в Италии и Аргентине подобная закономерность не выявлялась [8].
Клиническая картина
БУ является мультисистемным заболеванием, вследствие чего клиническая симптоматика чрезвычайно вариабельна. Заболевание начинается постепенно и в большинстве случаев носит хроническое течение со склонностью к рецидивам.
Наиболее ранним и очень часто (75%) единственным признаком БУ является суставной синдром, который может предшествовать развернутой клинической картине болезни в течение в среднем 6,7 лет. Имеется наблюдение, в котором интервал между поражением суставов и развитием желудочно–кишечных проявлений составил 36 лет [9].
Типичный случай поражения суставов был описан Уипплом, как «приступообразный артрит, захватывающий различные суставы и вовлекающий почти все суставы, пораженные к этому моменту времени. Эти приступы были преходящими, первый длился около 8 часов. Они рецидивировали вновь и вновь до 3–4 раз в неделю, в сырую погоду, продолжались от 6 до 24 часов, реже они были выраженными и не позволяли больному работать. Иногда суставы были припухшими и болезненными, реже – только болезненными» [1]. Действительно, у большинства больных отмечается приступообразный мигрирующий олиго– или полиартрит (реже – моноартрит), длящийся от нескольких часов до нескольких дней и отличающийся различной частотой обострений и полной ремиссией между приступами. Поражаются преимущественно коленные, лучезапястные и голеностопные суставы (рис. 1). В большинстве случаев артрит неэрозивный, протекает без развития деформаций, ревматоидный фактор не выявляется. Возможно развитие подкожных узелков, сходных с ревматоидными, однако при гистологическом исследовании в них выявлялись PAS– позитивные макрофаги, что подтверждало диагноз БУ. У отдельных больных отмечаются деструктивные изменения суставов. Описаны случаи формирования анкилоза лучезапястных и голеностопных суставов, а также гипертрофическая остеоартропатия. В 40% случаев встречаются спондилоартропатии, которые, как правило, сочетаются с периферическим артритом. Одно– или двухсторонний сакроилеит обнаруживают у 14% больных. Имеются описания анкилозирования крестцово–подвздошных сочленений, развития дерматомиозита и иных миопатий.
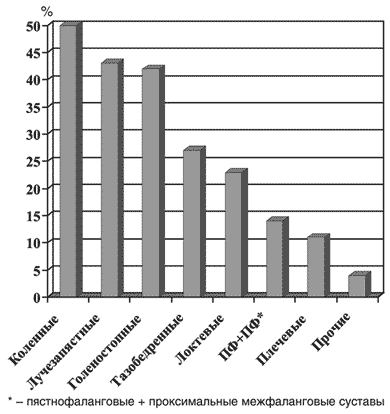
Рис. 1. Поражение суставов при БУ (n=131) [10]
Классическими признаками заболевания являются желудочно–кишечные расстройства. В типичных случаях отмечается диарея (чаще – стеаторея), сопровождающаяся приступообразной болью в животе, синдром нарушенного всасывания и прогрессирующая потеря массы тела. Как следствие, развивается полигиповитаминоз и разнообразные нарушения водно–электролитного обмена.Примерно у половины больных в развернутой стадии заболевания наблюдается лихорадочный синдром, сопровождающийся ознобом с последующим профузным потоотделением, а также генерализованная лимфаденопатия. Отмечается сухость кожи и ее диффузная гиперпигментация, преимущественно, на открытых участках тела (но не на слизистых), хейлит, глоссит, ломкость ногтей, аллопеция, отеки. Описано развитие пурпуры (без тромбоцитопении).
При БУ нередко встречается стойкий кашель, плеврит и пневмонит.
Клинические симптомы поражения сердечно–сосудистой системы наблюдаются в 30–55% случаев. Возможно поражение любой из оболочек сердца, однако наиболее часто развивается эндокардит. Последний по сравнению с инфекционным эндокардитом, вызванным другими возбудителями, имеет ряд особенностей: отсутствие предшествовавшей клапанной патологии сердца, нормальная температура тела, отрицательная гемокультура, позитивные ЭхоКГ– данные отмечаются только в 75% случаев. [11]. Описано несколько случаев эндокардита при БУ без гастроэнтерологической симптоматики [12,13]. Нередко развивается гипотензия и прогрессирующая застойная сердечная недостаточность.
Поражение ЦНС встречается в 10–50% случаев. Неврологическая симптоматика может быть как первым проявлением заболевания, так и развиваться спустя несколько лет на фоне манифестной клинической картины БУ. Наиболее частые симптомы – деменция, супрануклеарная офтальмоплегия и миоклония, которые встречаются в 25–50% случаев среди больных с патологией ЦНС при БУ. Также описаны эпилептические припадки, церебральная атаксия, инсомния. Сочетание медленного плавного конвергентно–дивергентного маятникообразного нистагма с синхронным сокращением жевательных мышц (oculomasticatory myorhytmia) или лицевых мышц (oculofaciosceetal myorhytmia) наблюдаются у 20% больных с вовлечением ЦНС и (несмотря на относительную редкость) рассматриваются как патогномоничные для БУ [14].
Наиболее распространенными формами поражения глаз при БУ являются увеит и офтальмоплегия. Встречается диффузный хориоретинит, глаукома, кератит.
В 10–15% случаев выявляют гепатомегалию, спленомегалию, асцит. Патология желудочно–кишечного тракта и эндокринной системы встречается крайне редко.
В течении БУ выделяют три стадии : I – стадия внекишечных симптомов (полиартрит, лихорадка), II – стадия мальабсорбции с прогрессирующим похуданием и тяжелыми метаболическими расстройствами, III – стадия системных проявлений (кардиальных, неврологических и т.д.).
Диагностика
Учитывая полиморфизм клинических проявлений, диагноз БУ весьма часто вызывает существенные трудности. Наибольшие диагностические проблемы возникают у больных с внекишечными формами болезни. По данным отечественных авторов, диагноз БУ устанавливается в среднем спустя 6 лет после первых клинических проявлений [15].
Какие–либо специфические изменения лабораторных показателей не наблюдаются. У больных значительно повышена СОЭ, а также число лейкоцитов и тромбоцитов, СРБ, снижен уровень гемоглобина, железа, кальция, калия, белка, альбуминов, холестерина (как следствие мальабсорбции). Отмечаются положительные результаты функциональных проб с ксилозой, нагрузкой глюкозой и др.
Важная роль в диагностике БУ отводится морфологическому исследованию слизистой оболочки тонкой кишки. При эндоскопии выявляют отек, гиперемию, и резкое утолщение складок кишки по причине лимфостаза, а также неровность рельефа слизистой из–за многочисленных желтовато–белых бляшек. При световой микроскопии биоптатов, полученных из тощей и 12–перстной кишки нелеченных больных с БУ, выявляют булавовидные ворсинки, содержащие большое количество лимфы. Часто отмечают внутри– и внеклеточное накопление жира в слизистой оболочке тонкой кишки. В собственной пластинке слизистой оболочки наблюдается большое количество пенистых макрофагов, содержащих крупнозернистые цитоплазматические включения с PAS–позитивной реакцией. Эти PAS–позитивные макрофаги могут также обнаруживаться в периферических или брыжеечных лимфоузлах, печени, селезенке, сердечных клапанах, мозговой ткани, стекловидном теле и синовиальной оболочке.
Считают, что PAS–позитивное вещество – это продукты распада фагоцитированных бактерий. Выявление PAS–позитивных включений в цитоплазме макрофагов не является патогномоничным для БУ (как полагали ранее), а может иметь место при других инфекциях, вызванных комплексом M. avium–intracellulare (у ВИЧ–инфицированных больных), коринебактериозе, гистоплазмозе, микозах, саркоидозе. В то же время PAS–реакция имеет чрезвычайно важное дифференциально–диагностическое значение, в частности, при разграничении БУ с первичной (болезнь Вальдмана) и приобретенными лимфангиэктазиями.
Ценным диагностическим подспорьем может служить электронная микроскопия, которая позволяет выявить в пораженных тканях палочковидные бактерии, локализующиеся как внутриклеточно, так и в межклеточном пространстве.
Несоблюдение правил проведения биопсии (взятие материала из верхней, а не из нижней части 12–перстной кишки) и предшествовавшая антимикробная терапия могут повлечь за собой отрицательные результаты патоморфологического исследования даже при характерной для БУ клинической картине. В подобных ситуациях особенно необходимой представляется ПЦР– диагностика. Использование праймеров, комплементарных консервативным участкам рибосомных генов (16S– и 23S–РНК), и дальнейшее определение полной нуклеотидной последовательности амплифицированной ДНК позволяют с высокой степенью вероятности идентифицировать T. whippelii. Для выполнения ПЦР могут быть использованы биоптаты слизистой 12–перстной кишки, лимфоузлов, сердечных клапанов (удаленных во время кардиохирургической операции), а также пунктаты синовиальной жидкости или ликвора. Несмотря на высокую чувствительность и специфичность ПЦР, существует определенная вероятность получения как ложноположительных, так и ложноотрицательных результатов. Поэтому залогом успешной диагностики БУ (как и любого другого заболевания) является комплексная оценка клинической картины и данных дополнительных исследований.
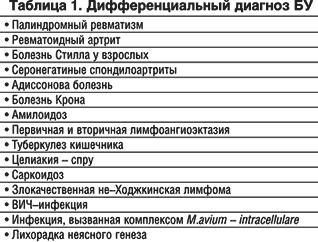
Дифференциальный диагноз БУ проводят с заболеваниями, имеющими сходную клиническую симптоматику (табл. 1). Необходимо заметить, что БУ обязательно должна быть включена в алгоритм диагностического поиска при лихорадке неясного генеза.
В доантибиотическую эру БУ заканчивалась летально в 100% наблюдений. Начиная с 1952 г., когда было сообщено об успешном применении антимикробного препарата (хлорамфеникола) для лечения БУ, до сегодняшнего дня продолжаются поиски оптимальных схем антибактериальной терапии. К сожалению, редкая встречаемость БУ, а также проблемы, связанные с культурированием T. whippelii, служат труднопреодолимым препятствием для проведения рандомизированных клинических исследований. Поэтому существующие схемы антимикробной терапии (табл. 2) разработаны на эмпирической основе.
В доантибиотическую эру БУ заканчивалась летально в 100% наблюдений. Начиная с 1952 г., когда было сообщено об успешном применении антимикробного препарата (хлорамфеникола) для лечения БУ, до сегодняшнего дня продолжаются поиски оптимальных схем антибактериальной терапии. К сожалению, редкая встречаемость БУ, а также проблемы, связанные с культурированием , служат труднопреодолимым препятствием для проведения рандомизированных клинических исследований. Поэтому существующие схемы антимикробной терапии (табл. 2) разработаны на эмпирической основе.
По мнению большинства авторов, при БУ, протекающей с неврологической симптоматикой, лечение следует начинать с 2–недельного парентерального введения бактерицидных антибиотиков, обладающих хорошей пенетрацией в спинномозговую жидкость, с последующим длительным (1–2 года) поддерживающим лечением ко– тримоксазолом. В остальных случаях терапия начинается с ко–тримоксазола, что считается более успешным в плане профилактики неврологических осложнений.
Многие авторы отмечают выраженную положительную динамику при своевременно начатой и активной терапии. Так, лихорадка и диарея купируются к концу 1–й недели лечения, суставной синдром полностью регрессирует в течение 1 месяца. В эти же сроки отмечается явное улучшение общего самочувствия, прибавка в весе. Обратное развитие неврологической симптоматики происходит значительно медленнее.
Вспомогательное значение имеет применение глюкокортикостероидов (преднизолон 30–40 мг в сутки с постепенным снижением дозы вплоть до полной отмены).
При необходимости в план лечения включают мероприятия, направленные на ликвидацию последствий синдрома мальабсорбции (коррекция метаболических нарушений, водно–электролитного обмена, восполнение дефицита железа и витаминов и т.д.)
Длительность антибактериальной терапии должна составлять не менее 1 года. Контроль за лечением осуществляют с помощью повторных морфологических исследований биоптатов тонкого кишечника либо ПЦР – методик. При отсутствии ответа на антибактериальную терапию представляется перспективным применение g–интерферона.
Прогноз в большинстве случаев благоприятный. При возникновении рецидивов (8–35%) терапия аналогична вышеуказанной.
Наиболее неблагоприятными в прогностическом плане считают неврологические осложнения, возникающие во время рецидивов, как следствие первично нераспознанной локализации инфекции или неадекватной терапии.
1. Whipple G.H. A hitherto undescribed disease characterized anatomically by deposits of fat and fatty acids in the intestinal mesenteric lymphatic issues. //John Hopkins Hospital Bulletin. 1907; 18: 382–391.
2. Black–Schaffer B. The tinctorial demonstration of a glycoprotein in Whipple’s disease. //Proc. Soc. Exp. Biol. Med. 1949; 72: 225 – 227.
3. Gardley J., Hendrix T.R. Combined electron and light microscopy in Whipple’s disease: demonstration of «bacillary bones» in the intestine. Bull. Johns Hopkins Hosp. 1961; 10: 80 –98.
4. Wilson K.H., Blitchington R.,Frothingham R., Wilson J.A.Phylogeny of the Whipple’s disease – associated bacterium.// Lancet. 1991; 338: 474–475.
5. Relman D.A.,Schmidt T.M., Mac Dermott R.P., Falkow S. Identification of the uncultured bacillus of the Whipple’s disease. N. Engl. J. Med. 1992; 327: 293–301.
6. Raoult D., Birq M.C., LaScola B., et al. Cultivation of the bacillus of Whipple’s disease. N. Engl. J. Med. 2000; 342: 620 – 625.
7. Dobbins W.O., Ruffin J.M.A light– and electron – microscopic study of bacterial invasion in Whipple’s disease. Am. J. Pathol. 1967; 51: 225 – 242.
8. Marth T., Raoult D. Whipple’s disease. Lancet. 2003; 361: 239 – 246.
9. Ayoub W.T., Davis D.E., Torreri D., Viozzi F.J. Bone destruction and ankylosis in Whipple’s disease. J. Rheumatol. 1982; 9: 930–931.
10. Puechal X. Whipple’s disease. Joint Bone Spine. 2002; 68: 133 – 140.
11. Fenollar F, Lepidi H, Raoult D. Whipple’s endocarditis: review of the literature and comparisons with Q fever, bartonella infection, and blood culture–positive endocarditis. Clin Infect Dis 2001;33(8):1309–1316.
12. Gubler JG, Kuster M, Dutly F, Bannwart F, Krause M, Vogelin HP, Garzoli G, Altwegg M. Whipple endocarditis without overt gastrointestinal disease: report of four cases. Ann. Intern. Med. 1999; 131(2):112–116.
13. Richardson DC, Burrows LL, Korithoski B, et al. Tropheryma whippelii as a cause of afebrile culture–negative endocarditis: the evolving spectrum of Whipple’s disease. J. Infect. 2003;47(2):170–173.
14. Ratnaike R.W. Whipple’s disease. Postgrad. Med. J. 2000; 76: 760 – 766.
15. Логинов А.С., Парфенов А.И., Полева Н.И. Болезнь Уиппла: результаты длительного наблюдения. Тер. арх. 1998; 9: 35 –41.
16. Misbah S.A., Mapstone N.P. Whipple’s disease revisited. J. Clin. Pathol. 2000; 53: 750 – 755.
17. Singer R. Diagnosis and treatment of Whipple’s disease (review). Drugs. 1998; 55: 699 – 704.



