




–Ш—Б—В–Њ—А–Є—П –≤–Њ–њ—А–Њ—Б–∞. –Я–µ—А–≤—Л–µ –Њ–њ–Є—Б–∞–љ–Є—П –і–µ—А–Љ–∞—В–Њ–Љ–Є–Њ–Ј–Є—В–∞ –Є –њ–Њ–ї–Є–Љ–Є–Њ–Ј–Є—В–∞ –њ—А–Є–љ–∞–і–ї–µ–ґ–∞—В –љ–µ–Љ–µ—Ж–Ї–Є–Љ –Ї–ї–Є–љ–Є—Ж–Є—Б—В–∞–Љ –≤ –њ–µ—А–Є–Њ–і –Љ–µ–ґ–і—Г 1886 –Є 1891 –≥. –Ґ–µ—А–Љ–Є–љ «–њ–Њ–ї–Є–Љ–Є–Њ–Ј–Є—В» –њ—А–µ–і–ї–Њ–ґ–µ–љ E. Wagner –≤ 1886 –≥–Њ–і—Г, –∞ –≤ 1891 –≥. – H. Unverricht –Њ–±—А–∞—В–Є–ї –≤–љ–Є–Љ–∞–љ–Є–µ –љ–∞ —Б–Њ—З–µ—В–∞–љ–Є–µ –Ї–Њ–ґ–љ–Њ–≥–Њ –Є –Љ—Л—И–µ—З–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–ї —В–µ—А–Љ–Є–љ «–і–µ—А–Љ–∞—В–Њ–Љ–Є–Њ–Ј–Є—В». –Ю–Ї–Њ–ї–Њ 2/3 –Њ–њ–Є—Б–∞–љ–Є–є XIX –≤–µ–Ї–∞ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—В –њ–Њ–ї–Є–Љ–Є–Њ–Ј–Є—В—Г, –Њ—Б—В–∞–≤—И–Є–µ—Б—П – –і–µ—А–Љ–∞—В–Њ–Љ–Є–Њ–Ј–Є—В—Г. –Т 1916 –≥–Њ–і—Г –Њ–њ–Є—Б–∞–љ–∞ –∞—Б—Б–Њ—Ж–Є–∞—Ж–Є—П —Б–Њ –Ј–ї–Њ–Ї–∞—З–µ—Б—В–≤–µ–љ–љ—Л–Љ–Є –Њ–њ—Г—Е–Њ–ї—П–Љ–Є, –Њ–і–љ–∞–Ї–Њ –њ—А–Є—З–Є–љ–љ–∞—П –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј—М –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–∞ —В–Њ–ї—М–Ї–Њ –≤ 1935 –≥–Њ–і—Г. –Т 1967 Chou –≤–њ–µ—А–≤—Л–µ –Њ–њ–Є—Б–∞–ї –Љ–Є–Њ–Ј–Є—В —Б –≤–Ї–ї—О—З–µ–љ–Є—П–Љ–Є, —Е–Њ—В—П —Б–∞–Љ —В–µ—А–Љ–Є–љ «–Љ–Є–Њ–Ј–Є—В —Б –≤–Ї–ї—О—З–µ–љ–Є—П–Љ–Є» –≤–≤–µ–і–µ–љ Yunis –Є Samaha –≤ 1971 –≥–Њ–і—Г, –Ї–Њ–≥–і–∞ –≤ –≥–Є—Б—В–Њ–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Б—А–µ–Ј–∞—Е –Љ—Л—И–µ—З–љ–Њ–є —В–Ї–∞–љ–Є –±—Л–ї–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ —П–і–µ—А–љ—Л–µ —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є–µ –≤–Ї–ї—О—З–µ–љ–Є—П [2].
–І–∞—Б—В–Њ—В–∞ –Я–Ь/–Ф–Ь –≤ –њ–Њ–њ—Г–ї—П—Ж–Є–Є –Ї–Њ–ї–µ–±–ї–µ—В—Б—П –Њ—В 2 –і–Њ 10 –љ–∞ 1 –Љ–ї–љ –љ–∞—Б–µ–ї–µ–љ–Є—П –≤ –≥–Њ–і. –Ф–Ь, —А–µ–ґ–µ –Я–Ь, –∞—Б—Б–Њ—Ж–Є–Є—А—Г—О—Й–Є–µ—Б—П —Б –Њ–њ—Г—Е–Њ–ї—П–Љ–Є, —Б–Њ—Б—В–∞–≤–ї—П—О—В –њ—А–Є–Љ–µ—А–љ–Њ 20% –Њ—В –≤—Б–µ—Е —Б–ї—Г—З–∞–µ–≤ –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Љ–Є–Њ–њ–∞—В–Є–є. –Ю–њ—Г—Е–Њ–ї–Є –Љ–Њ–≥—Г—В —А–∞–Ј–≤–Є–≤–∞—В—М—Б—П –і–Њ –њ–Њ—П–≤–ї–µ–љ–Є—П –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –Ш–Т–Ь, –Њ–і–љ–Њ–≤—А–µ–Љ–µ–љ–љ–Њ —Б –љ–Є–Љ–Є –Є–ї–Є –њ–Њ—Б–ї–µ –Є—Е –њ–Њ—П–≤–ї–µ–љ–Є—П. –І–∞—Б—В–Њ—В–∞ –Ј–ї–Њ–Ї–∞—З–µ—Б—В–≤–µ–љ–љ—Л—Е –љ–Њ–≤–Њ–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–є –њ—А–Є –Я–Ь/–Ф–Ь –≤ 12 —А–∞–Ј –≤—Л—И–µ, —З–µ–Љ –≤ –њ–Њ–њ—Г–ї—П—Ж–Є–Є. –Э–∞ —Д–Њ–љ–µ –Ј–ї–Њ–Ї–∞—З–µ—Б—В–≤–µ–љ–љ—Л—Е –љ–Њ–≤–Њ–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–є —З–∞—Й–µ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –Ф–Ь, —З–µ–Љ –Я–Ь [1,3].
–≠—В–Є–Њ–ї–Њ–≥–Є—П. –Э–∞ —А–Њ–ї—М –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–є –њ—А–µ–і—А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ–Њ—Б—В–Є —Г–Ї–∞–Ј—Л–≤–∞–µ—В –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В—М —А–∞–Ј–≤–Є—В–Є—П –Я–Ь/–Ф–Ь —Г –Љ–Њ–љ–Њ–Ј–Є–≥–Њ—В–љ—Л—Е –±–ї–Є–Ј–љ–µ—Ж–Њ–≤ –Є –Ї—А–Њ–≤–љ—Л—Е —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤ –њ–∞—Ж–Є–µ–љ—В–Њ–≤. –Э–Њ—Б–Є—В–µ–ї—М—Б—В–≤–Њ –љ–µ–Ї–Њ—В–Њ—А—Л—Е HLA (HLA––Т8/DR3, HLA B14 –Є HLA B40) –±–Њ–ї–µ–µ —В–µ—Б–љ–Њ —Б–≤—П–Ј–∞–љ–Њ –љ–µ —Б —Б–∞–Љ–Є–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ, –∞ —Б –Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л–Љ–Є –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є, –≤ –њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М –≥–Є–њ–µ—А–њ—А–Њ–і—Г–Ї—Ж–Є–µ–є –Љ–Є–Њ–Ј–Є—В–—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є—Е –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї (—В–∞–±–ї. 1) [1].
–Я–∞—В–Њ–≥–µ–љ–µ–Ј. –Т –Њ—Б–љ–Њ–≤–µ –њ–∞—В–Њ–≥–µ–љ–µ–Ј–∞ –Я–Ь/–Ф–Ь –ї–µ–ґ–∞—В –Ї–ї–µ—В–Њ—З–љ—Л–µ –Є –≥—Г–Љ–Њ—А–∞–ї—М–љ—Л–µ –Є–Љ–Љ—Г–љ–љ—Л–µ —А–µ–∞–Ї—Ж–Є–Є. –°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –≤ –њ–∞—В–Њ–≥–µ–љ–µ–Ј–µ –Я–Ь –Є –Ф–Ь —Б—Г—Й–µ—Б—В–≤—Г—О—В –њ—А–Є–љ—Ж–Є–њ–Є–∞–ї—М–љ—Л–µ –Є–Љ–Љ—Г–љ–Њ–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ —А–∞–Ј–ї–Є—З–Є—П.
–Я—А–Є –Ф–Ь —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –≥—Г–Љ–Њ—А–∞–ї—М–љ—Л–є –Є–Љ–Љ—Г–љ–љ—Л–є –Њ—В–≤–µ—В, –њ—А–Є–≤–Њ–і—П—Й–Є–є –Ї –∞–Ї—В–Є–≤–∞—Ж–Є–Є —Б–Є—Б—В–µ–Љ—Л –Ї–Њ–Љ–њ–ї–µ–Љ–µ–љ—В–∞ –Є —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—О –∞–Ї—В–Є–≤–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –°3, —З—В–Њ –≤–µ–і–µ—В –Ї —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—О C3b –љ–µ–Њ–∞–љ—В–Є–≥–µ–љ–∞ –Є –Љ–µ–Љ–±—А–∞–љ–Њ–ї–Є—В–Є—З–µ—Б–Ї–Њ–≥–Њ –∞—В–∞–Ї—Г—О—Й–µ–≥–Њ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞, –Ї–Њ—В–Њ—А—Л–є –Њ—В–Ї–ї–∞–і—Л–≤–∞–µ—В—Б—П –≤ —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Ї–∞—Е –Є –≤–Њ–Ї—А—Г–≥ –љ–Є—Е, –≤ —Б—В–µ–љ–Ї–µ —Н–љ–і–Њ–Љ–Є–Ј–Є–∞–ї—М–љ—Л—Е –Ї–∞–њ–Є–ї–ї—П—А–Њ–≤. –Ю—В–ї–Њ–ґ–µ–љ–Є–µ –Љ–µ–Љ–±—А–∞–љ–Њ–ї–Є—В–Є—З–µ—Б–Ї–Њ–≥–Њ –∞—В–∞–Ї—Г—О—Й–µ–≥–Њ –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –≤–µ–і–µ—В –Ї –і–µ—Б—В—А—Г–Ї—Ж–Є–Є –Є —Г–Љ–µ–љ—М—И–µ–љ–Є—О —З–Є—Б–ї–∞ –Ї–∞–њ–Є–ї–ї—П—А–Њ–≤ —Б –Є—И–µ–Љ–Є–µ–є –Є –Љ–Є–Ї—А–Њ–Є–љ—Д–∞—А–Ї—В–∞–Љ–Є –Љ–Є–Њ—Ж–Є—В–Њ–≤, –љ–∞–Є–±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ–љ–Њ–≥–Њ –љ–∞ –њ–µ—А–Є—Д–µ—А–Є–Є –њ—Г—З–Ї–∞. –Т —А–µ–Ј—Г–ї—М—В–∞—В–µ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –Ї–∞—А—В–Є–љ–∞ –Љ–∞–ї–Њ–≥–Њ —З–Є—Б–ї–∞ —Г–≤–µ–ї–Є—З–µ–љ–љ—Л—Е –≤ –і–Є–∞–Љ–µ—В—А–µ –Ї–∞–њ–Є–ї–ї—П—А–Њ–≤ –Є –њ–µ—А–Є—Д–∞—Б—Ж–Є–Ї—Г–ї—П—А–љ–∞—П –∞—В—А–Њ—Д–Є—П [3].
–Я—А–Є –Ф–Ь —В–∞–Ї–ґ–µ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –Љ–Є–≥—А–∞—Ж–Є—П –Т––Ї–ї–µ—В–Њ–Ї, CD4+ –Ґ––ї–Є–Љ—Д–Њ—Ж–Є—В–Њ–≤ –Є –Љ–∞–Ї—А–Њ—Д–∞–≥–Њ–≤, –Њ–±—А–∞–Ј—Г—О—Й–Є—Е –≤–љ—Г—В—А–Є–Љ—Л—И–µ—З–љ—Л–є –Є–љ—Д–Є–ї—М—В—А–∞—В. –Ь–Є–≥—А–∞—Ж–Є–Є —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В –≤–∞—Б–Ї—Г–ї—П—А–љ—Л–µ –Љ–Њ–ї–µ–Ї—Г–ї—Л –Ї–ї–µ—В–Њ—З–љ–Њ–є –∞–і–≥–µ–Ј–Є–Є 1 (VCAM1) –Є –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ—Л–µ –Љ–Њ–ї–µ–Ї—Г–ї—Л –∞–і–≥–µ–Ј–Є–Є 1 (ICAM1), —Н–Ї—Б–њ—А–µ—Б—Б–Є—П –Ї–Њ—В–Њ—А—Л—Е –љ–∞ —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ—Л—Е –Ї–ї–µ—В–Ї–∞—Е –њ–Њ–≤—Л—И–µ–љ–∞ –Ј–∞ —Б—З–µ—В –≤–Њ–Ј–і–µ–є—Б—В–≤–Є—П –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–љ—Л—Е —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤, —Б –њ–Њ—Б–ї–µ–і—Г—О—Й–Є–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –≤–љ—Г—В—А–Є–Љ—Л—И–µ—З–љ—Л—Е –Љ–Є–Ї—А–Њ—Б–Њ—Б—Г–і–Њ–≤ –Є —А–∞–Ј–≤–Є—В–Є–µ–Љ –≤–љ—Г—В—А–Є–Љ—Л—И–µ—З–љ–Њ–є –≤–∞—Б–Ї—Г–ї–Њ–њ–∞—В–Є–Є. –¶–Є—В–Њ–Ї–Є–љ—Л, –≤—Л—Б–≤–Њ–±–Њ–ґ–і–µ–љ–љ—Л–µ –∞–Ї—В–Є–≤–Є—А–Њ–≤–∞–љ–љ—Л–Љ–Є –Ґ– –Є –Т––Ї–ї–µ—В–Ї–∞–Љ–Є, —Г—Б–Є–ї–Є–≤–∞—О—В –њ—А–Њ—Ж–µ—Б—Б —В—А–∞–љ—Б–Љ–Є–≥—А–∞—Ж–Є–Є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –≤ –Љ—Л—И—Ж—Г [3].
–Я—А–Є –Я–Ь –Є –Љ–Є–Њ–Ј–Є—В–µ —Б –≤–Ї–ї—О—З–µ–љ–Є—П–Љ–Є —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В—Б—П –Ї–ї–Њ–љ –∞—Г—В–Њ–Є–љ–≤–∞–Ј–Є–≤–љ—Л—Е –°D8+ –Ґ––Ї–ї–µ—В–Њ–Ї. CD8+ –Ґ––ї–Є–Љ—Д–Њ—Ж–Є—В—Л —Б–Є–љ—В–µ–Ј–Є—А—Г—О—В —Ж–Є—В–Њ—В–Њ–Ї—Б–Є—З–µ—Б–Ї–Є–µ —Б—Г–±—Б—В–∞–љ—Ж–Є–Є (–њ–µ—А—Д–Њ—А–Є–љ, –≥—А–∞–љ–Ј–Є–Љ) –Є —З–µ—А–µ–Ј –њ–µ—А—Д–Њ—А–Є–љ–Њ–≤—Л–є –њ—Г—В—М –Њ–Ї–∞–Ј—Л–≤–∞—О—В –Љ–Є–Њ—В–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–µ –і–µ–є—Б—В–≤–Є–µ –љ–∞ –Љ–Є–Њ—Д–Є–±—А–Є–ї–ї—Л, —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г—О—Й–Є–µ –Љ–Њ–ї–µ–Ї—Г–ї—Л HLA–I –Ї–ї–∞—Б—Б–∞ [1–4]. –Т –і–µ–±—О—В–µ –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞ –Я–Ь –Є –Љ–Є–Њ–Ј–Є—В–∞ —Б –≤–Ї–ї—О—З–µ–љ–Є—П–Љ–Є –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ–Њ–≤—Л—И–µ–љ–Є–µ —Н–Ї—Б–њ—А–µ—Б—Б–Є–Є –Љ–Њ–ї–µ–Ї—Г–ї HLA–I –љ–∞ –Љ—Л—И–µ—З–љ—Л—Е –≤–Њ–ї–Њ–Ї–љ–∞—Е, –і–∞–ґ–µ –≤ –Њ—В—Б—Г—В—Б—В–≤–Є–µ –∞—Г—В–Њ–Є–љ–≤–∞–Ј–Є–≤–љ—Л—Е –°D8+ –Ґ––Ї–ї–µ—В–Њ–Ї [3].
–Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –±–Њ–ї–µ–Ј–љ—М –і–µ–±—О—В–Є—А—Г–µ—В –љ–µ–і–Њ–Љ–Њ–≥–∞–љ–Є–µ–Љ, –Њ–±—Й–µ–є —Б–ї–∞–±–Њ—Б—В—М—О, –Љ–Є–∞–ї–≥–Є—П–Љ–Є, –њ—А–µ—Е–Њ–і—П—Й–Є–Љ —Б–Є–Љ–Љ–µ—В—А–Є—З–љ—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —Б—Г—Б—В–∞–≤–Њ–≤, –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –Ї–Њ–ґ–Є. –Э–∞—З–∞–ї—Г –±–Њ–ї–µ–Ј–љ–Є –Љ–Њ–ґ–µ—В –њ—А–µ–і—И–µ—Б—В–≤–Њ–≤–∞—В—М –Є–љ—Б–Њ–ї—П—Ж–Є—П. –Ч–∞—В–µ–Љ, –≤ —В–µ—З–µ–љ–Є–µ –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е –љ–µ–і–µ–ї—М (–Љ–µ—Б—П—Ж–µ–≤), –њ–Њ—Б—В–µ–њ–µ–љ–љ–Њ –љ–∞—А–∞—Б—В–∞–µ—В —Б–ї–∞–±–Њ—Б—В—М –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –≥—А—Г–њ–њ –Љ—Л—И—Ж. –£ –і–µ—В–µ–є –Є –ї–Є—Ж –Љ–Њ–ї–Њ–і–Њ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞ –Љ–Њ–ґ–µ—В –љ–∞–±–ї—О–і–∞—В—М—Б—П –±–Њ–ї–µ–µ –Њ—Б—В—А–Њ–µ –љ–∞—З–∞–ї–Њ, —З–∞—Б—В–Њ —Б–Њ—З–µ—В–∞—О—Й–µ–µ—Б—П —Б –≤—Л—А–∞–ґ–µ–љ–љ—Л–Љ–Є –Ї–Њ–љ—Б—В–Є—В—Г—Ж–Є–Њ–љ–∞–ї—М–љ—Л–Љ–Є –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є (–ї–Є—Е–Њ—А–∞–і–Ї–∞, –њ–Њ—Е—Г–і–∞–љ–Є–µ –Є –і—А.). –£ –±–Њ–ї—М–љ—Л—Е —Б –∞–Љ–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–Љ –Ф–Ь –≤ —В–µ—З–µ–љ–Є–µ –і–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ –≤—А–µ–Љ–µ–љ–Є –Љ–Њ–ґ–µ—В –љ–∞–±–ї—О–і–∞—В—М—Б—П —В–Є–њ–Є—З–љ–Њ–µ –і–ї—П –Ф–Ь –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Ї–Њ–ґ–Є –њ—А–Є –Њ—В—Б—Г—В—Б—В–≤–Є–Є –Љ—Л—И–µ—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В–Є [1].
–Т–µ–і—Г—Й–Є–Љ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П [5,6] —П–≤–ї—П–µ—В—Б—П –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Љ—Л—И—Ж, –≤—Л—А–∞–ґ–∞—О—Й–µ–µ—Б—П —Б–Є–Љ–Љ–µ—В—А–Є—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В—М—О –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –≥—А—Г–њ–њ –Љ—Л—И—Ж –≤–µ—А—Е–љ–Є—Е –Є –љ–Є–ґ–љ–Є—Е –Ї–Њ–љ–µ—З–љ–Њ—Б—В–µ–є –Є –Љ—Л—И—Ж —И–µ–Є, –≤–µ–і—Г—Й–µ–є –Ї –Ј–∞—В—А—Г–і–љ–µ–љ–Є—О –њ—А–Є –њ–Њ–і—К–µ–Љ–µ —Б –љ–Є–Ј–Ї–Њ–≥–Њ —Б—В—Г–ї–∞, –њ–Њ—Б–∞–і–Ї–µ –≤ —В—А–∞–љ—Б–њ–Њ—А—В, –њ—А–Є —Г–Љ—Л–≤–∞–љ–Є–Є –Є –њ—А–Є—З–µ—Б—Л–≤–∞–љ–Є–Є; –±–Њ–ї—М–љ–Њ–є –љ–µ –Љ–Њ–ґ–µ—В –Њ—В–Њ—А–≤–∞—В—М –≥–Њ–ї–Њ–≤—Г –Њ—В –њ–Њ–і—Г—И–Ї–Є. –Э–∞–±–ї—О–і–∞—О—В—Б—П: –Є–Ј–Љ–µ–љ–µ–љ–Є–µ –њ–Њ—Е–Њ–і–Ї–Є –Є —Н–њ–Є–Ј–Њ–і—Л –љ–µ–Њ–ґ–Є–і–∞–љ–љ—Л—Е –њ–∞–і–µ–љ–Є–є. –Ь–Њ–ґ–µ—В —А–∞–Ј–≤–Є–≤–∞—В—М—Б—П –Њ—В–µ–Ї –Љ—Л—И—Ж. –•–∞—А–∞–Ї—В–µ—А–љ–Њ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Љ—Л—И—Ж –≥–ї–Њ—В–Ї–Є, –≥–Њ—А—В–∞–љ–Є –Є –≤–µ—А—Е–љ–µ–є —В—А–µ—В–Є –њ–Є—Й–µ–≤–Њ–і–∞ (–і–Є—Б—Д–Њ–љ–Є—П, –і–Є—Б—Д–∞–≥–Є—П). –Р–Љ–Є–Њ—В—А–Њ—Д–Є–Є —А–∞–Ј–≤–Є–≤–∞—О—В—Б—П —В–Њ–ї—М–Ї–Њ —Г –±–Њ–ї—М–љ—Л—Е, –і–ї–Є—В–µ–ї—М–љ–Њ —Б—В—А–∞–і–∞—О—Й–Є—Е –Я–Ь/–Ф–Ь.
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –і–Є—Б—В–∞–ї—М–љ–Њ–є –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А—Л –љ–µ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ –Є –љ–∞–±–ї—О–і–∞–µ—В—Б—П, –≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ, –њ—А–Є –Љ–Є–Њ–Ј–Є—В–µ —Б «–≤–Ї–ї—О—З–µ–љ–Є—П–Љ–Є».
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –Ї–Њ–ґ–Є –њ—А–Є –Ф–Ь —З–∞—Б—В–Њ –њ—А–µ–і—И–µ—Б—В–≤—Г–µ—В —А–∞–Ј–≤–Є—В–Є—О –Љ—Л—И–µ—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В–Є. –•–∞—А–∞–Ї—В–µ—А–љ—Л–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ —П–≤–ї—П–µ—В—Б—П —Н—А–Є—В–µ–Љ–∞—В–Њ–Ј–љ–∞—П («–≥–µ–ї–Є–Њ—В—А–Њ–њ–љ–∞—П») —Б—Л–њ—М, –ї–Њ–Ї–∞–ї–Є–Ј—Г—О—Й–∞—П—Б—П –љ–∞ –≤–µ—А—Е–љ–Є—Е –≤–µ–Ї–∞—Е, —Б–Ї—Г–ї–∞—Е, –≤ –Ј–Њ–љ–µ «–і–µ–Ї–Њ–ї—М—В–µ» –Є «—И–∞–ї–Є» (—А–Є—Б. 1), –љ–∞–і –ї–Њ–Ї—В–µ–≤—Л–Љ–Є, –Ї–Њ–ї–µ–љ–љ—Л–Љ–Є, –њ—П—Б—В–љ–Њ—Д–∞–ї–∞–љ–≥–Њ–≤—Л–Љ–Є –Є –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л–Љ–Є –Љ–µ–ґ—Д–∞–ї–∞–љ–≥–Њ–≤—Л–Љ–Є —Б—Г—Б—В–∞–≤–∞–Љ–Є – —Н—А–Є—В–µ–Љ–∞/–њ–∞–њ—Г–ї—Л –У–Њ—В—В—А–Њ–љ–∞ (—А–Є—Б. 2), —Н—А–Є—В–µ–Љ–∞ –≤–Њ–ї–Њ—Б–Є—Б—В–Њ–є —З–∞—Б—В–Є –≥–Њ–ї–Њ–≤—Л (—А–Є—Б. 3). –Ґ–∞–Ї–ґ–µ –љ–∞–±–ї—О–і–∞—О—В—Б—П —И–µ–ї—Г—И–µ–љ–Є–µ –Є —В—А–µ—Й–Є–љ—Л –љ–∞ –Ї–Њ–ґ–µ –ї–∞–і–Њ–љ–µ–є («—А—Г–Ї–∞ –Љ–µ—Е–∞–љ–Є–Ї–∞»), –Њ–Ї–Њ–ї–Њ–љ–Њ–≥—В–µ–≤–∞—П —Н—А–Є—В–µ–Љ–∞, —Д–Њ—В–Њ–і–µ—А–Љ–∞—В–Є—В, –Ї–Њ–ґ–љ—Л–є –Ј—Г–і. –Ь–Њ–≥—Г—В –љ–∞–±–ї—О–і–∞—В—М—Б—П —В–∞–Ї–Є–µ —Д–Њ—А–Љ—Л —Б–Њ—Б—Г–і–Є—Б—В–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є, –Ї–∞–Ї –Є–љ—Д–∞—А–Ї—В—Л –Њ–Ї–Њ–ї–Њ–љ–Њ–≥—В–µ–≤–Њ–≥–Њ –ї–Њ–ґ–∞, –њ–µ—В–µ—Е–Є–Є –Є —Б–µ—В—З–∞—В–Њ–µ –ї–Є–≤–µ–і–Њ [6,7].
–Р—А—В—А–Є—В—Л/–∞—А—В—А–∞–ї–≥–Є–Є –љ–∞–±–ї—О–і–∞—О—В—Б—П –і–Њ—Б—В–∞—В–Њ—З–љ–Њ —А–µ–і–Ї–Њ –Є –±—Л—Б—В—А–Њ –Ї—Г–њ–Є—А—Г—О—В—Б—П –њ—А–Є –љ–∞–Ј–љ–∞—З–µ–љ–Є–Є –≥–ї—О–Ї–Њ–Ї–Њ—А—В–Є–Ї–Њ–Є–і–Њ–≤ (–У–Ъ). –†–∞–Ј–≤–Є—В–Є–µ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –і–µ—Д–Њ—А–Љ–Є—А—Г—О—Й–µ–≥–Њ –∞—А—В—А–Є—В–∞ —Б –њ–Њ–і–≤—Л–≤–Є—Е–∞–Љ–Є —Б—Г—Б—В–∞–≤–Њ–≤ –Ї–Є—Б—В–µ–є –љ–∞–±–ї—О–і–∞–µ—В—Б—П —А–µ–і–Ї–Њ –Є –љ–µ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П —Н—А–Њ–Ј–Є–≤–љ—Л–Љ–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є (—Б–Є–љ–і—А–Њ–Љ –Ц–∞–Ї—Г) (—А–Є—Б. 4) [1].
–Ъ–∞–ї—М—Ж–Є–љ–Њ–Ј –Љ—П–≥–Ї–Є—Е —В–Ї–∞–љ–µ–є –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–Є –Ѓ–Ф–Ь, –њ—А–Є –њ–µ—А–µ–Ї—А–µ—Б—В–љ—Л—Е —Б–Є–љ–і—А–Њ–Љ–∞—Е –Љ–Є–Њ–Ј–Є—В–∞ (–љ–∞–њ—А–Є–Љ–µ—А, —Б —Б–Є—Б—В–µ–Љ–љ–Њ–є —Б–Ї–ї–µ—А–Њ–і–µ—А–Љ–Є–µ–є) –Є–ї–Є –љ–∞ –њ–Њ–Ј–і–љ–Є—Е —Б—В–∞–і–Є—П—Е –Я–Ь/–Ф–Ь. –Ъ–∞–ї—М—Ж–Є–љ–∞—В—Л —А–∞—Б–њ–Њ–ї–∞–≥–∞—О—В—Б—П –њ–Њ–і–Ї–Њ–ґ–љ–Њ, –≤ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є, –≤–Њ–Ї—А—Г–≥ –Љ—Л—И–µ—З–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ –Є, –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ, –≤ –Ј–Њ–љ–∞—Е —В—А–∞–≤–Љ–∞—В–Є–Ј–∞—Ж–Є–Є –Є–ї–Є –њ–Њ—Б–ї–µ –≤–љ—Г—В—А–Є–Љ—Л—И–µ—З–љ—Л—Е –Є–љ—К–µ–Ї—Ж–Є–є, –љ–∞–њ—А–Є–Љ–µ—А, –љ–∞ —П–≥–Њ–і–Є—Ж–∞—Е [1].
–Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –≤—Б—В—А–µ—З–∞—О—Й–Є–Љ—Б—П –Њ—А–≥–∞–љ–љ—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —П–≤–ї—П–µ—В—Б—П –њ–Њ—А–∞–ґ–µ–љ–Є–µ –і—Л—Е–∞—В–µ–ї—М–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л (80%) [8,9]. –Я—А–Є –Я–Ь/–Ф–Ь –љ–∞–±–ї—О–і–∞—О—В—Б—П —А–∞–Ј–ї–Є—З–љ—Л–µ –≤–∞—А–Є–∞–љ—В—Л –њ–Њ—А–∞–ґ–µ–љ–Є—П –і—Л—Е–∞—В–µ–ї—М–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л.
–£ –њ–Њ–і–∞–≤–ї—П—О—Й–µ–≥–Њ –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –≤–Њ–≤–ї–µ—З–µ–љ–Є–µ –≤ –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є –њ—А–Њ—Ж–µ—Б—Б –Ї–∞–Ї –Љ–µ–ґ—А–µ–±–µ—А–љ—Л—Е –Љ—Л—И—Ж, —В–∞–Ї –Є –і–Є–∞—Д—А–∞–≥–Љ—Л. –Т—Л—Б–Њ–Ї–Њ–µ —Б—В–Њ—П–љ–Є–µ –Ї—Г–њ–Њ–ї–Њ–≤ –і–Є–∞—Д—А–∞–≥–Љ—Л –Є –≤—П–ї–Њ—Б—В—М –µ–µ –і—Л—Е–∞—В–µ–ї—М–љ–Њ–є —Н–Ї—Б–Ї—Г—А—Б–Є–Є –њ—А–Є–≤–Њ–і–Є—В –Ї —Н–Ї—Б–њ–Є—А–∞—В–Њ—А–љ–Њ–є –Њ–і—Л—И–Ї–µ, –Є–Ј–Љ–µ–љ–µ–љ–Є—О —Д—Г–љ–Ї—Ж–Є–Є –≤–љ–µ—И–љ–µ–≥–Њ –і—Л—Е–∞–љ–Є—П –њ–Њ —А–µ—Б—В—А–Є–Ї—В–Є–≤–љ–Њ–Љ—Г —В–Є–њ—Г. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –љ–∞—А—Г—И–µ–љ–Є–µ –≥–ї–Њ—В–∞–љ–Є—П, —Б–≤—П–Ј–∞–љ–љ–Њ–µ —Б –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –≥–ї–Њ—В–Њ—З–љ—Л—Е –Љ—Л—И—Ж, –Љ–Њ–ґ–µ—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї –∞—Б–њ–Є—А–∞—Ж–Є–Є –њ–Є—Й–Є –Є —Б–ї—О–љ—Л —Б –њ–Њ—Б–ї–µ–і—Г—О—Й–Є–Љ —А–∞–Ј–≤–Є—В–Є–µ–Љ –∞—Б–њ–Є—А–∞—Ж–Є–Њ–љ–љ–Њ–є –њ–љ–µ–≤–Љ–Њ–љ–Є–Є.
–Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ–є —Д–Њ—А–Љ–Њ–є –ї–µ–≥–Њ—З–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П —П–≤–ї—П–µ—В—Б—П –њ–љ–µ–≤–Љ–Њ–љ–Є—П, –Ї–Њ—В–Њ—А–∞—П –≤—Б—В—А–µ—З–∞–µ—В—Б—П –њ—А–Є –Я–Ь/–Ф–Ь –≤ 29–54% —Б–ї—Г—З–∞–µ–≤. –Т–µ–і—Г—Й—Г—О —А–Њ–ї—М –≤ –µ–µ —А–∞–Ј–≤–Є—В–Є–Є –Є–≥—А–∞–µ—В –∞—Б–њ–Є—А–∞—Ж–Є—П –њ–Є—Й–Є. –†–Є—Б–Ї —А–∞–Ј–≤–Є—В–Є—П –њ–љ–µ–≤–Љ–Њ–љ–Є–є –Є —В—А—Г–і–љ–Њ—Б—В–Є –њ—А–Є –Є—Е –ї–µ—З–µ–љ–Є–Є –≤–Њ–Ј—А–∞—Б—В–∞—О—В –≤ —Б–≤—П–Ј–Є –≤—Л—А–∞–ґ–µ–љ–љ—Л–Љ –Є–Љ–Љ—Г–љ–Њ–і–µ—Д–Є—Ж–Є—В–Њ–Љ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Я–Ь/–Ф–Ь, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–Љ –і–ї–Є—В–µ–ї—М–љ—Л–Љ –њ—А–Є–µ–Љ–Њ–Љ –≤—Л—Б–Њ–Ї–Є—Е –і–Њ–Ј –У–Ъ –Є –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є–≤–љ–Њ–є —В–µ—А–∞–њ–Є–Є. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –≥–Є–њ–Њ–≤–µ–љ—В–Є–ї—П—Ж–Є–Њ–љ–љ—Л–є —Б–Є–љ–і—А–Њ–Љ —Г—Б—Г–≥—Г–±–ї—П–µ—В —А–Є—Б–Ї —А–∞–Ј–≤–Є—В–Є—П –њ–љ–µ–≤–Љ–Њ–љ–Є–Є [8].
–Э–∞–Є–±–Њ–ї–µ–µ —В—П–ґ–µ–ї—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –ї–µ–≥–Њ—З–љ–Њ–є —В–Ї–∞–љ–Є —П–≤–ї—П–µ—В—Б—П —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–Є–є –∞–ї—М–≤–µ–Њ–ї–Є—В, –Ї–Њ—В–Њ—А—Л–є –≤ –Ј–∞—А—Г–±–µ–ґ–љ–Њ–є –ї–Є—В–µ—А–∞—В—Г—А–µ –њ—А–Є–љ—П—В–Њ –љ–∞–Ј—Л–≤–∞—В—М –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ –ї–µ–≥–Ї–Є—Е (–Ш–Ч–Ы). –§–Є–±—А–Њ–Ј–Є—А—Г—О—Й–Є–є –∞–ї—М–≤–µ–Њ–ї–Є—В –њ—А–Є –Я–Ь/–Ф–Ь –Є–Љ–µ–µ—В —И–Є—А–Њ–Ї–Є–є —Б–њ–µ–Ї—В—А –Ї–ї–Є–љ–Є–Ї–Њ––ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є –Є —Б—Е–Њ–і–µ–љ —Б –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–Љ —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–Є–Љ –∞–ї—М–≤–µ–Њ–ї–Є—В–Њ–Љ. Targoff I.N. c —Б–Њ–∞–≤—В.1992, –≤—Л–і–µ–ї—П—О—В —В—А–Є —Д–Њ—А–Љ—Л —Б–Є–љ–і—А–Њ–Љ–∞ —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–µ–≥–Њ –∞–ї—М–≤–µ–Њ–ї–Є—В–∞: 1 – –Я–Њ —В–Є–њ—Г –±—Л—Б—В—А–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞ —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–µ–≥–Њ –∞–ї—М–≤–µ–Њ–ї–Є—В–∞ (—Б–Є–љ–і—А–Њ–Љ –•–∞–Љ–Љ–∞–љ–∞––†–Є—З–Є): –Њ—Б—В—А—Л–є –љ–µ–њ—А–Њ–і—Г–Ї—В–Є–≤–љ—Л–є –Ї–∞—И–µ–ї—М, –Њ–і—Л—И–Ї–∞ –≤ –њ–Њ–Ї–Њ–µ, –ї–Є—Е–Њ—А–∞–і–Ї–∞. –Э–∞ —А–µ–љ—В–≥–µ–љ–Њ–≥—А–∞–Љ–Љ–∞—Е – –Љ–љ–Њ–ґ–µ—Б—В–≤–µ–љ–љ—Л–µ –Љ–µ–ї–Ї–Њ–Њ—З–∞–≥–Њ–≤—Л–µ –Ј–∞—В–µ–Љ–љ–µ–љ–Є—П –Є —Б–µ—В—З–∞—В–∞—П –і–µ—Д–Њ—А–Љ–∞—Ж–Є—П –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —А–Є—Б—Г–љ–Ї–∞. –°–Є–Љ–њ—В–Њ–Љ—Л –њ–Њ—А–∞–ґ–µ–љ–Є—П –Љ—Г—Б–Ї—Г–ї–∞—В—Г—А—Л –Љ–Њ–≥—Г—В –±—Л—В—М –љ–∞ –≤—В–Њ—А–Њ–Љ –њ–ї–∞–љ–µ. –Я—А–Њ–≥–љ–Њ–Ј –≤ –і–∞–љ–љ–Њ–Љ —Б–ї—Г—З–∞–µ, –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–є. 2 – –С–Њ–ї–µ–µ –Љ–µ–і–ї–µ–љ–љ–Њ–µ —А–∞–Ј–≤–Є—В–Є–µ –±–Њ–ї–µ–Ј–љ–Є, –≤ –і–µ–±—О—В–µ: –Њ–і—Л—И–Ї–∞ –њ—А–Є —Д–Є–Ј–Є—З–µ—Б–Ї–Њ–є –љ–∞–≥—А—Г–Ј–Ї–µ –Є/–Є–ї–Є –љ–µ–њ—А–Њ–і—Г–Ї—В–Є–≤–љ—Л–є –Ї–∞—И–µ–ї—М. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Љ–Њ–≥—Г—В –њ—А–µ–і—И–µ—Б—В–≤–Њ–≤–∞—В—М –Љ—Л—И–µ—З–љ–Њ–Љ—Г –њ–Њ—А–∞–ґ–µ–љ–Є—О, –≤–Њ–Ј–љ–Є–Ї–∞—В—М –Њ–і–љ–Њ–≤—А–µ–Љ–µ–љ–љ–Њ —Б –љ–Є–Љ –Є–ї–Є —А–∞–Ј–≤–Є–≤–∞—В—М—Б—П –љ–∞ —Д–Њ–љ–µ –Є–Љ–µ—О—Й–µ–≥–Њ—Б—П —В—П–ґ–µ–ї–Њ–≥–Њ –Љ–Є–Њ–Ј–Є—В–∞. –£ —А—П–і–∞ –±–Њ–ї—М–љ—Л—Е 1 –Є 2 –≥—А—Г–њ–њ –Љ–Њ–ґ–µ—В –њ—А–Є—Б—Г—В—Б—В–≤–Њ–≤–∞—В—М —В–∞—Е–Є–њ–љ–Њ—Н, –Ї—А–µ–њ–Є—В–∞—Ж–Є—П –≤ –љ–Є–ґ–љ–Є—Е –Њ—В–і–µ–ї–∞—Е –ї–µ–≥–Ї–Є—Е, —А–µ–ґ–µ —А–∞–Ј–≤–Є—В–Є–µ –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —Б–µ—А–і—Ж–∞. 3 – –°—Г–±–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ: —П—А–Ї–∞—П –Ї–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –ї–µ–≥–Њ—З–љ–∞—П —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–∞ –Њ—В—Б—Г—В—Б—В–≤—Г–µ—В. –Ш–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤ –ї–µ–≥–Њ—З–љ–Њ–є —В–Ї–∞–љ–Є –≤—Л—П–≤–ї—П—О—В—Б—П —В–Њ–ї—М–Ї–Њ –њ—А–Є –њ–Њ–Љ–Њ—Й–Є —Б–њ–µ—Ж–Є–∞–ї—М–љ—Л—Е –Љ–µ—В–Њ–і–Њ–≤ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П (–Ъ–Ґ, —А–µ–љ—В–≥–µ–љ–Њ–≥—А–∞—Д–Є—П) [8].
–Ґ–∞–Ї, –Ї–ї–Є–љ–Є–Ї–∞ —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–µ–≥–Њ –∞–ї—М–≤–µ–Њ–ї–Є—В–∞ –Љ–Њ–ґ–µ—В –Њ–њ–µ—А–µ–ґ–∞—В—М —А–∞–Ј–≤–Є—В–Є–µ –Љ—Л—И–µ—З–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞ –Є–ї–Є –њ—А–Њ—П–≤–ї—П—В—М—Б—П –Њ–і–љ–Њ–≤—А–µ–Љ–µ–љ–љ–Њ —Б –љ–Є–Љ. –Ю–і–љ–∞–Ї–Њ –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є —Б–Є–љ–і—А–Њ–Љ–∞ —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–µ–≥–Њ –∞–ї—М–≤–µ–Њ–ї–Є—В–∞ —А–∞–Ј–≤–Є–≤–∞—О—В—Б—П –љ–∞ —Д–Њ–љ–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ—Л –Я–Ь/–Ф–Ь, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –љ–∞ —А–∞–љ–љ–Є—Е —Н—В–∞–њ–∞—Е –±–Њ–ї–µ–Ј–љ–Є.
–Я–Њ—А–∞–ґ–µ–љ–Є–µ —Б–µ—А–і—Ж–∞ –≤ –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ –њ—А–Њ—В–µ–Ї–∞–µ—В –±–µ–Ј —П–≤–љ—Л—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П. –Т–µ–і—Г—Й–µ–µ –Љ–µ—Б—В–Њ –≤ —Б—В—А—Г–Ї—В—Г—А–µ –Ї–∞—А–і–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є –њ—А–Є –Ш–Т–Ь –Ј–∞–љ–Є–Љ–∞–µ—В –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Љ–Є–Њ–Ї–∞—А–і–∞, –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –њ—А–Њ—П–≤–ї—П—О—Й–µ–µ—Б—П –љ–∞—А—Г—И–µ–љ–Є–µ–Љ —А–Є—В–Љ–∞ (—Б—Г–њ—А–∞–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–∞—П –Є–ї–Є –ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤–∞—П —Н–Ї—Б—В—А–∞—Б–Є—Б—В–Њ–ї–Є—П) –Є –њ—А–Њ–≤–Њ–і–Є–Љ–Њ—Б—В–Є (–љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ – –±–ї–Њ–Ї–∞–і–∞ –ї–µ–≤–Њ–є –љ–Њ–ґ–Ї–Є –њ—Г—З–Ї–∞ –У–Є—Б–∞), –∞ —В–∞–Ї–ґ–µ —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –µ–≥–Њ —Б–Њ–Ї—А–∞—В–Є—В–µ–ї—М–љ–Њ–є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є. –Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є–µ–Љ –љ–∞—А—Г—И–µ–љ–Є—П —Б–Њ–Ї—А–∞—В–Є—В–µ–ї—М–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є —Б–µ—А–і—Ж–∞ —П–≤–ї—П–µ—В—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ –ї–Њ–Ї–∞–ї—М–љ–Њ–є —Б–Њ–Ї—А–∞—В–Є–Љ–Њ—Б—В–Є –≤ –Њ–±–ї–∞—Б—В–Є –Ј–∞–і–љ–µ–є —Б—В–µ–љ–Ї–Є –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –Є –Љ–µ–ґ–ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤–Њ–є –њ–µ—А–µ–≥–Њ—А–Њ–і–Ї–Є. –Ъ—А–∞–є–љ–µ —А–µ–і–Ї–Њ —А–∞–Ј–≤–Є–≤–∞—О—В—Б—П –Љ–Є–Њ–Ї–∞—А–і–Є—В –Є–ї–Є –Ј–∞—Б—В–Њ–є–љ–∞—П —Б–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М –љ–∞ —Д–Њ–љ–µ –Љ–Є–Њ–Ї–∞—А–і–Є–Њ—Д–Є–±—А–Њ–Ј–∞ [10].
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –Ц–Ъ–Ґ: –љ–∞—А—Г—И–µ–љ–Є–µ –≥–ї–Њ—В–∞–љ–Є—П –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –Љ—Л—И—Ж –≥–ї–Њ—В–Ї–Є, –≥–Є–њ–Њ—В–Њ–љ–Є—П –њ–Є—Й–µ–≤–Њ–і–∞ (–≤–µ—А—Е–љ—П—П —В—А–µ—В—М) [1]. –Я–Њ—А–∞–ґ–µ–љ–Є–µ –њ–Њ—З–µ–Ї –љ–∞–±–ї—О–і–∞–µ—В—Б—П –Ї—А–∞–є–љ–µ —А–µ–і–Ї–Њ, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –њ—А–Є overlap–—Б–Є–љ–і—А–Њ–Љ–∞—Е, –≤ –Њ—Б–љ–Њ–≤–љ–Њ–Љ —Б –°–°–Ф. –Т–Њ–Ј–Љ–Њ–ґ–љ–∞ –њ—А–Њ—В–µ–Є–љ—Г—А–Є—П. –Ю–њ–Є—Б–∞–љ—Л –µ–і–Є–љ–Є—З–љ—Л–µ —Б–ї—Г—З–∞–Є —А–∞–Ј–≤–Є—В–Є—П —А–∞–±–і–Њ–Љ–Є–Њ–ї–Є–Ј–∞ (–Љ–Є–Њ–≥–ї–Њ–±–Є–љ—Г—А–Є—П – –Њ—Б—В—А–∞—П –њ–Њ—З–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М) [1,4].
–§–µ–љ–Њ–Љ–µ–љ –†–µ–є–љ–Њ —З–∞—Й–µ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–Є –Ф–Ь, –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ–Њ–Љ —Б–Є–љ–і—А–Њ–Љ–µ –Є —Г –±–Њ–ї—М–љ—Л—Е —Б –њ–µ—А–µ–Ї—А–µ—Б—В–љ—Л–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ –Я–Ь/–Ф–Ь –Є —Б–Њ —Б–Љ–µ—И–∞–љ–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є [1,4,11].
–Ъ–ї–Є–љ–Є–Ї–Њ––Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –∞—Б—Б–Њ—Ж–Є–∞—Ж–Є–Є (—В–∞–±–ї. 1). –°–Њ–≥–ї–∞—Б–љ–Њ —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л–Љ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Є—П–Љ, —А–∞–Ј–≤–Є—В–Є–µ –Ш–Т–Ь —Б–≤—П–Ј–∞–љ–Њ —Б —Б–Є–љ—В–µ–Ј–Њ–Љ —Ж–µ–ї–Њ–≥–Њ «—Б–µ–Љ–µ–є—Б—В–≤–∞» –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л—Е –Ї —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є–Љ –±–µ–ї–Ї–∞–Љ –Є —А–Є–±–Њ–љ—Г–Ї–ї–µ–Є–љ–Њ–≤—Л–Љ –Ї–Є—Б–ї–Њ—В–∞–Љ. –≠—В–Є –∞–љ—В–Є—В–µ–ї–∞ –њ—А–Є—Б—Г—В—Б—В–≤—Г—О—В –≤ —Б—Л–≤–Њ—А–Њ—В–Ї–∞—Е —Г –њ–Њ—З—В–Є 90% –±–Њ–ї—М–љ—Л—Е, —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞—О—В—Б—П –Ї–∞–Ї –Љ–Є–Њ–Ј–Є—В—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–µ –∞–љ—В–Є—В–µ–ї–∞ –Є –њ–Њ–і—А–∞–Ј–і–µ–ї—П—О—В—Б—П –љ–∞ –њ–Њ–і–≥—А—Г–њ–њ—Л [4,8]. –Т—Л—П–≤–ї–µ–љ–Є–µ –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї –Ї–∞–ґ–і–Њ–є –њ–Њ–і–≥—А—Г–њ–њ—Л –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–Њ —Б –Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л–Љ —Б–Є–Љ–њ—В–Њ–Љ–Њ–Ї–Њ–Љ–њ–ї–µ–Ї—Б–Њ–Љ. –Э–∞–Є–±–Њ–ї–µ–µ —В—П–ґ–µ–ї—Л–Љ —П–≤–ї—П–µ—В—Б—П «–∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–є» —Б–Є–љ–і—А–Њ–Љ, –Ї–Њ—В–Њ—А—Л–є –Љ–∞—А–Ї–Є—А—Г—О—В –∞–љ—В–Є—В–µ–ї–∞ –Ї –∞–Љ–Є–љ–Њ–∞—Ж–Є–ї—Б–Є–љ—В–µ—В–∞–Ј–∞–Љ —В–†–Э–Ъ (Jo–1–Є –і—А.) [11].
–Ы–∞–±–Њ—А–∞—В–Њ—А–љ—Л–µ –і–∞–љ–љ—Л–µ –≤–Ї–ї—О—З–∞—О—В –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ–Њ–є –Ї—А–µ–∞—В–Є–љ—Д–Њ—Б—Д–Њ–Ї–Є–љ–∞–Ј—Л (–Ъ–§–Ъ), –ї–∞–Ї—В–∞—В–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј—Л (–Ы–Ф–У), –∞—Б–њ–∞—А–∞–≥–Є–љ–Њ–≤–Њ–є (–Р–°–Ґ), –∞–ї–∞–љ–Є–љ–Њ–≤–Њ–є (–Р–Ы–Ґ) —В—А–∞–љ—Б–∞–Љ–Є–љ–∞–Ј. –Я—А–Є –Љ—Л—И–µ—З–љ–Њ–Љ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–Є –Љ–Њ–ґ–µ—В –љ–∞–±–ї—О–і–∞—В—М—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П —Б—Л–≤–Њ—А–Њ—В–Њ—З–љ–Њ–≥–Њ –Ї—А–µ–∞—В–Є–љ–Є–љ–∞ [11–13].
–Р–Э–§ –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П —Г 50–80%, —З–∞—Й–µ —Г –±–Њ–ї—М–љ—Л—Е —Б –њ–µ—А–µ–Ї—А–µ—Б—В–љ—Л–Љ–Є —Б–Є–љ–і—А–Њ–Љ–∞–Љ–Є –Љ–Є–Њ–Ј–Є—В–∞ —Б –і—А—Г–≥–Є–Љ–Є –°–Ч–°–Ґ –Є –≤ –±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Є—Е —В–Є—В—А–∞—Е [14]. –Ю–њ—А–µ–і–µ–ї–µ–љ–Є–µ –∞–љ—В–Є—В–µ–ї –Ї –∞–Љ–Є–љ–Њ–∞—Ж–Є–ї—Б–Є–љ—В–µ—В–∞–Ј–∞–Љ —В—А–∞–љ—Б–њ–Њ—А—В–љ–Њ–є –†–Э–Ъ, –≤ –њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М Jo–1: –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–є –Ї—А–Є—В–µ—А–Є–є –Я–Ь/–Ф–Ь –Є –ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л–є –Љ–∞—А–Ї–µ—А «–∞–љ—В–Є—Б–Є–љ—В–∞–Ј–љ–Њ–≥–Њ» —Б–Є–љ–і—А–Њ–Љ–∞ [15–18].
–≠–ї–µ–Ї—В—А–Њ–Љ–Є–Њ–≥—А–∞—Д–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Є–≥–Њ–ї—М—З–∞—В—Л–Љ–Є —Н–ї–µ–Ї—В—А–Њ–і–∞–Љ–Є (–Є––≠–Ь–У) –њ—А–Њ–≤–Њ–і–Є—В—Б—П —Б —Ж–µ–ї—М—О –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є—П –њ–µ—А–≤–Є—З–љ–Њ––Љ—Л—И–µ—З–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П, –Њ–њ—А–µ–і–µ–ї–µ–љ–Є—П —Б—В–µ–њ–µ–љ–Є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞ –Є –љ–µ–Ї—А–Њ–Ј–∞ –Љ—Л—И–µ—З–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ. –Я—А–Є –Я–Ь/–Ф–Ь –љ–∞–±–ї—О–і–∞–µ—В—Б—П —Б–љ–Є–ґ–µ–љ–Є–µ –∞–Љ–њ–ї–Є—В—Г–і—Л –Є —Б—А–µ–і–љ–µ–є –і–ї–Є—В–µ–ї—М–љ–Њ—Б—В–Є –њ–Њ—В–µ–љ—Ж–Є–∞–ї–Њ–≤ –і–≤–Є–≥–∞—В–µ–ї—М–љ—Л—Е –µ–і–Є–љ–Є—Ж (–Я–Ф–Х), –њ–Њ–ї–Є—Д–∞–Ј–Є—П, –њ—Б–µ–≤–і–Њ–њ–Њ–ї–Є—Д–∞–Ј–Є—П –Є —Б–њ–Њ–љ—В–∞–љ–љ–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М (–°–Р) –Љ—Л—И–µ—З–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ (–њ–Њ—В–µ–љ—Ж–Є–∞–ї—Л —Д–Є–±—А–Є–ї–ї—П—Ж–Є–є –Є –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–µ –Њ—Б—В—А—Л–µ –≤–Њ–ї–љ—Л). –Я–Њ–≤–µ–і–µ–љ–Є–µ –Є––≠–Ь–У –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Є –≤ –і–Є–љ–∞–Љ–Є–Ї–µ, –і–ї—П –Њ—Ж–µ–љ–Ї–Є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ—А–Њ–≤–Њ–і–Є–Љ–Њ–є —В–µ—А–∞–њ–Є–Є [19–20].
–°–Њ–≥–ї–∞—Б–љ–Њ –Ї–ї–∞—Б—Б–Є—З–µ—Б–Ї–Є–Љ –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–Љ –Ї—А–Є—В–µ—А–Є—П–Љ, –≤ –Љ—Л—И–µ—З–љ–Њ–Љ –±–Є–Њ–њ—В–∞—В–µ –≤—Л—П–≤–ї—П—О—В—Б—П: –љ–µ–Ї—А–Њ–Ј –Є –∞—В—А–Њ—Д–Є—П –Љ—Л—И–µ—З–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ, –≤—Л—А–∞–ґ–µ–љ–љ–∞—П –ї–Є–Љ—Д–Њ–≥–Є—Б—В–Є–Њ—Ж–Є—В–∞—А–љ–∞—П –Є–љ—Д–Є–ї—М—В—А–∞—Ж–Є—П, —Г–Љ–µ—А–µ–љ–љ–∞—П —А–µ–≥–µ–љ–µ—А–∞—Ж–Є—П –Љ—Л—И–µ—З–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ, –њ–Њ—В–µ—А—П –њ–Њ–њ–µ—А–µ—З–љ–Њ–њ–Њ–ї–Њ—Б–∞—В–Њ–є –Є—Б—З–µ—А—З–µ–љ–љ–Њ—Б—В–Є [12,24].
–Ъ–∞–њ–Є–ї–ї—П—А–Њ—Б–Ї–Њ–њ–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–∞ –і–µ—Б—В—А—Г–Ї—Ж–Є–µ–є –Є –і–µ–Ј–Њ—А–≥–∞–љ–Є–Ј–∞—Ж–Є–µ–є –Ї–∞–њ–Є–ї–ї—П—А–Њ–≤, —Г–Љ–µ–љ—М—И–µ–љ–Є–µ–Љ –Є—Е —З–Є—Б–ї–∞, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ —А–∞–Ј–Љ–µ—А–∞; –љ–µ–Њ–∞–љ–≥–Є–Њ–≥–µ–љ–µ–Ј–Њ–Љ, –∞ —В–∞–Ї–ґ–µ —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ–Љ —В–∞–Ї –љ–∞–Ј—Л–≤–∞–µ–Љ—Л—Е «–Ї—Г—Б—В–Њ–≤–Є–і–љ—Л—Е» –Ї–∞–њ–Є–ї–ї—П—А–Њ–≤ (—А–Є—Б. 5).
–†–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –ї–µ–≥–Ї–Є—Е –Є–ї–Є —А–µ–љ—В–≥–µ–љ–Њ–≤—Б–Ї–∞—П –Ї–Њ–Љ–њ—М—О—В–µ—А–љ–∞—П —В–Њ–Љ–Њ–≥—А–∞—Д–Є—П —Б –≤—Л—Б–Њ–Ї–Є–Љ —А–∞–Ј—А–µ—И–µ–љ–Є–µ–Љ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –≤—Л—П–≤–ї–µ–љ–Є—О –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –ї–µ–≥–Њ—З–љ–Њ–є —В–Ї–∞–љ–Є, –Њ—В –±–∞–Ј–∞–ї—М–љ–Њ–≥–Њ –њ–љ–µ–≤–Љ–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞ –і–Њ –Њ—Б—В—А–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–µ–≥–Њ –∞–ї—М–≤–µ–Њ–ї–Є—В–∞ [8].
–Т –њ–Њ—Б–ї–µ–і–љ–Є–µ –≥–Њ–і—Л –Љ–љ–Њ–≥–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–Є –Њ–њ–Є—А–∞—О—В—Б—П –љ–∞ —А–µ–Ј—Г–ї—М—В–∞—В—Л –Љ–∞–≥–љ–Є—В–љ–Њ–—А–µ–Ј–Њ–љ–∞–љ—Б–љ–Њ–є —В–Њ–Љ–Њ–≥—А–∞—Д–Є–Є (–Ь–†–Ґ) [22]. –Ь–†–Ґ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ—А–Њ–≤–µ—Б—В–Є —А–∞–љ–љ—О—О –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї—Г –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –±–ї–∞–≥–Њ–і–∞—А—П –≤—Л—П–≤–ї–µ–љ–Є—О –Њ—В–µ–Ї–∞ –Љ—Л—И–µ—З–љ–Њ–є —В–Ї–∞–љ–Є –і–∞–ґ–µ –і–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є –њ–Њ—А–∞–ґ–µ–љ–Є—П –Љ—Л—И—Ж, –Њ—Б–Њ–±–µ–љ–љ–Њ –≤ —Б–ї—Г—З–∞–µ –і–µ—А–Љ–∞—В–Њ–Љ–Є–Њ–Ј–Є—В–∞, –Ї–Њ–≥–і–∞ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Ї–Њ–ґ–љ—Л—Е –њ–Њ–Ї—А–Њ–≤–Њ–≤ —Г–ґ–µ –Њ—З–µ–≤–Є–і–љ–Њ. –Ю—В–µ–Ї –Љ—Л—И–µ—З–љ–Њ–є —В–Ї–∞–љ–Є —П–≤–ї—П–µ—В—Б—П –Є–љ–і–Є–Ї–∞—В–Њ—А–Њ–Љ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –±–Њ–ї–µ–Ј–љ–Є.
–Ч–∞ –≥–Њ–і—Л –Є–Ј—Г—З–µ–љ–Є—П –Ш–Т–Ь –љ–µ–Њ–і–љ–Њ–Ї—А–∞—В–љ–Њ –њ–Њ–≤—В–Њ—А—П–ї–Є—Б—М –њ–Њ–њ—Л—В–Ї–Є —А–∞–Ј—А–∞–±–Њ—В–Ї–Є –Њ–њ—В–Є–Љ–∞–ї—М–љ—Л—Е –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є—Е –Ї—А–Є—В–µ—А–Є–µ–≤ –Ш–Т–Ь: Bohan and Peter 1975; Dalakas 1991; Griggs 1995; Tanimoto 1995; Targoff 1997; Mastaglia 2002; Van der Meulen 2003; Dalakas, Hohlfeld 2003 [5–13]. –Э–∞–Є–±–Њ–ї–µ–µ —И–Є—А–Њ–Ї–Њ –≤ –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –Є—Б–њ–Њ–ї—М–Ј—Г—О—В—Б—П –Ї—А–Є—В–µ—А–Є–Є Bohan and Peter 1975 –Є Tanimoto 1995 (—В–∞–±–ї. 2) [12,13,23–28].
–Т—Л–Ј—Л–≤–∞—О—В –Є–љ—В–µ—А–µ—Б –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–µ –Ї—А–Є—В–µ—А–Є–Є, –њ—А–µ–і–ї–Њ–ґ–µ–љ–љ—Л–µ Dalakas –≤ 1991 –≥–Њ–і—Г, –Ї–Њ—В–Њ—А—Л–µ –Њ—Б–љ–Њ–≤–∞–љ—Л –љ–∞ —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є—Е –Є–Љ–Љ—Г–љ–Њ–≥–Є—Б—В–Њ–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—П—Е –Љ—Л—И–µ—З–љ–Њ–≥–Њ –Є–љ—Д–Є–ї—М—В—А–∞—В–∞ –Є –њ–Њ–Ј–≤–Њ–ї—П—О—В –і–Є—Д—Д–µ—А–µ–љ—Ж–Є—А–Њ–≤–∞—В—М –Ф–Ь –Є –Я–Ь –Њ—В –і—А—Г–≥–Є—Е –Љ–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є—Е —Б–Є–љ–і—А–Њ–Љ–Њ–≤ (—Б–Љ. –і–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ–∞—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞). –Ґ–∞–Ї, –њ–Њ –Љ–љ–µ–љ–Є—О Dalakas, –Љ—Л—И–µ—З–љ—Л–є –±–Є–Њ–њ—В–∞—В –њ—А–Є –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ–Љ –Я–Ь –Є–Љ–µ–µ—В –Є–љ–≤–∞–Ј–Є—О CD8+ –ї–Є–Љ—Д–Њ—Ж–Є—В–∞–Љ–Є –љ–µ–љ–µ–Ї—А–Њ—В–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е –Љ—Л—И–µ—З–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ, —Н–Ї—Б–њ—А–µ—Б—Б–Є—А—Г—О—Й–Є—Е HLA–I –Ї–ї–∞—Б—Б–∞ –≥–Є—Б—В–Њ—Б–Њ–≤–Љ–µ—Б—В–Є–Љ–Њ—Б—В–Є. –Я—А–Є —Н—В–Њ–Љ –Њ—В—Б—Г—В—Б—В–≤—Г—О—В –≤–∞–Ї—Г–Њ–ї–Є, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –і–ї—П –Љ–Є–Њ–Ј–Є—В–∞ —Б «–≤–Ї–ї—О—З–µ–љ–Є—П–Љ–Є». –Ф–Њ—Б—В–Њ–≤–µ—А–љ—Л–є –і–Є–∞–≥–љ–Њ–Ј –Ф–Ь –њ–Њ Dalakas —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –љ–∞–ї–Є—З–Є–µ–Љ –њ–µ—А–Є—Д–∞—Б—Ж–Є–∞–ї—М–љ–Њ–є –∞—В—А–Њ—Д–Є–Є –Є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Є–љ—Д–Є–ї—М—В—А–∞—В–Њ–≤ –≤ –Љ—Л—И—Ж–∞—Е –≤ —Б–Њ—З–µ—В–∞–љ–Є–Є —Б —Е–∞—А–∞–Ї—В–µ—А–љ—Л–Љ–Є –Ї–Њ–ґ–љ—Л–Љ–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є [21].
–Ф–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ–∞—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞ [19–20,29] –Ш–Т–Ь –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–ї–Њ–ґ–љ—Г—О –њ—А–Њ–±–ї–µ–Љ—Г –Є –њ—А–Њ–≤–Њ–і–Є—В—Б—П —Б —И–Є—А–Њ–Ї–Є–Љ —Б–њ–µ–Ї—В—А–Њ–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—Й–Є—Е—Б—П —В–∞–Ї–ґ–µ –Љ—Л—И–µ—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В—М—О, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ–Љ –Ъ–§–Ъ –Є –њ–µ—А–≤–Є—З–љ–Њ––Љ—Л—И–µ—З–љ—Л–Љ–Є —Н–ї–µ–Ї—В—А–Њ–Љ–Є–Њ–≥—А–∞—Д–Є—З–µ—Б–Ї–Є–Љ–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є.
–Ш—Б—В–Є–љ–љ—Г—О –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Г—О –Љ—Л—И–µ—З–љ—Г—О —Б–ї–∞–±–Њ—Б—В—М —Б–ї–µ–і—Г–µ—В –Њ—В–ї–Є—З–∞—В—М –Њ—В –Њ–±—Й–µ–є —Б–ї–∞–±–Њ—Б—В–Є, —Г—В–Њ–Љ–ї—П–µ–Љ–Њ—Б—В–Є –Є–ї–Є –Љ–Є–∞–ї–≥–Є–Є, –≤—Л–Ј—Л–≤–∞–µ–Љ—Л–Љ–Є —А–∞–Ј–ї–Є—З–љ—Л–Љ–Є –њ—А–Є—З–Є–љ–∞–Љ–Є.
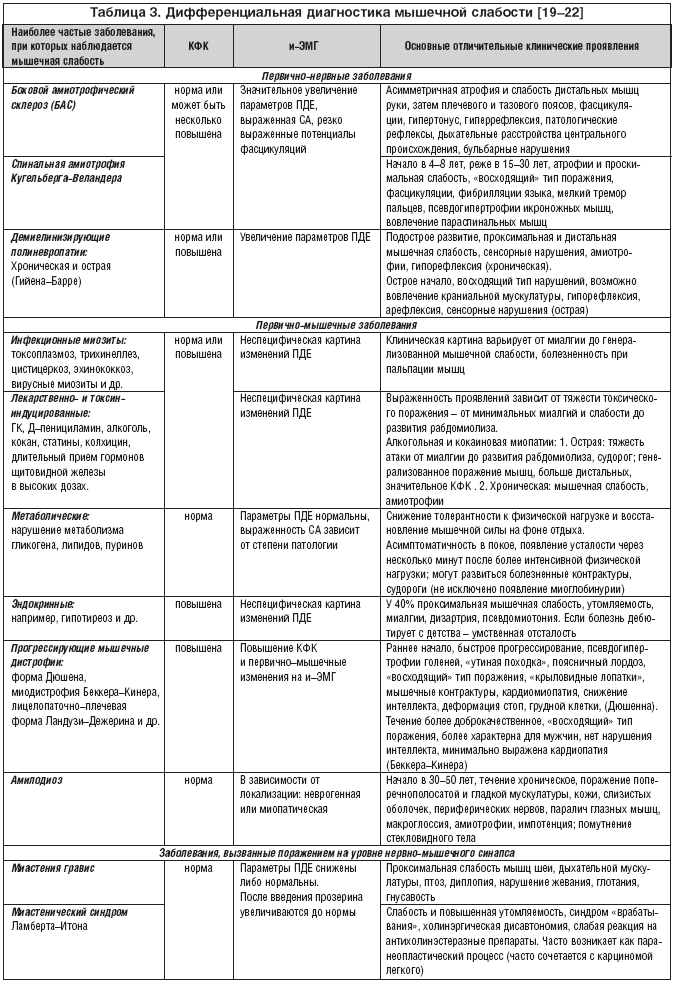
–£—З–Є—В—Л–≤–∞—П –∞–Ї—В—Г–∞–ї—М–љ–Њ—Б—В—М –Є —Б–ї–Њ–ґ–љ–Њ—Б—В—М –њ—А–Њ–±–ї–µ–Љ—Л –і–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ–Њ–є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Љ–Є–Њ–њ–∞—В–Є–є, –µ—Й–µ –≤ 1975 –≥–Њ–і—Г Bohan and Peter –±—Л–ї–Є –≤—Л–і–µ–ї–µ–љ—Л –Ї—А–Є—В–µ—А–Є–Є –Є—Б–Ї–ї—О—З–µ–љ–Є—П [12], –Њ–±—К–µ–і–Є–љ—П—О—Й–Є–µ —А–∞–Ј–ї–Є—З–љ—Л–µ –љ–Њ–Ј–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ —Д–Њ—А–Љ—Л, –≥–і–µ –Њ–і–љ–Є–Љ –Є–Ј –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є –Љ–Њ–ґ–µ—В —П–≤–ї—П—В—М—Б—П –Љ—Л—И–µ—З–љ–∞—П —Б–ї–∞–±–Њ—Б—В—М. –Т –њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М, —Н—В–Њ –љ–µ–≤—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –Љ–Є–∞—Б—В–µ–љ–Є—П –≥—А–∞–≤–Є—Б –Є –Љ–Є–Њ–і–Є—Б—В—А–Њ—Д–Є–Є, –∞ —В–∞–Ї–ґ–µ —Б–∞—А–Ї–Њ–Є–і–Њ–Ј, –Є–љ—Д–µ–Ї—Ж–Є–Њ–љ–љ—Л–µ –±–Њ–ї–µ–Ј–љ–Є (—В—А–Є—Е–µ–љ–µ–ї–ї–µ–Ј, —Ж–Є—Б—В–µ—А–Ї–Њ–Ј, —В–Њ–Ї—Б–Њ–њ–ї–∞–Ј–Љ–Њ–Ј), —В–Њ–Ї—Б–Є–љ– –Є –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ–Њ––Є–љ–і—Г—Ж–Є—А–Њ–≤–∞–љ–љ—Л–µ –Љ–Є–Њ–њ–∞—В–Є–Є, —А–∞–±–і–Њ–Љ–Є–Њ–ї–Є–Ј, –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–µ –Љ–Є–Њ–њ–∞—В–Є–Є (–љ–∞–њ—А–Є–Љ–µ—А, –±–Њ–ї–µ–Ј–љ—М –Ь–∞–Ї –Р—А–і–ї–∞) –Є —Н–љ–і–Њ–Ї—А–Є–љ–Њ–њ–∞—В–Є–Є (—В–∞–±–ї. 3).
–Т 2000 –≥. —Б–Њ–Ј–і–∞–љ–∞ –Љ–µ–ґ–і—Г–љ–∞—А–Њ–і–љ–∞—П –Љ—Г–ї—М—В–Є–і–Є—Б—Ж–Є–њ–ї–Є–љ–∞—А–љ–∞—П –≥—А—Г–њ–њ–∞ –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–µ–є –њ–Њ –Њ—Ж–µ–љ–Ї–µ –Љ–Є–Њ–Ј–Є—В–∞ –Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є (International Myositis Assessment and Clinical Studies Group – IMACS) –њ–Њ–і —А—Г–Ї–Њ–≤–Њ–і—Б—В–≤–Њ–Љ –њ—А–Њ—Д–µ—Б—Б–Њ—А–∞ P. Plotz. –†–∞–Ј—А–∞–±–Њ—В–∞–љ—Л —Б—В–∞–љ–і–∞—А—В–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л–µ –Љ–µ—В–Њ–і—Л –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –±–Њ–ї—М–љ—Л—Е –Ш–Т–Ь (—В–∞–±–ї. 4) [30–32].
–Я—А–µ–і–ї–Њ–ґ–µ–љ—Л —Б–ї–µ–і—Г—О—Й–Є–µ –і–µ—Д–Є–љ–Є—Ж–Є–Є:
–Р–Ї—В–Є–≤–љ–Њ—Б—В—М —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–µ—В—Б—П –Ї–∞–Ї —З–∞—Б—В–Є—З–љ–Њ –Є–ї–Є –њ–Њ–ї–љ–Њ—Б—В—М—О –Њ–±—А–∞—В–Є–Љ—Л–µ —П–≤–ї–µ–љ–Є—П, —Б–≤—П–Ј–∞–љ–љ—Л–µ –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ —Б –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–Љ –њ—А–Њ—Ж–µ—Б—Б–Њ–Љ –≤ –Љ—Л—И—Ж–∞—Е. –Я–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ – –Ї—А–Є—В–µ—А–Є–є –Є—Б—Е–Њ–і–∞ –±–Њ–ї–µ–Ј–љ–Є, –Њ—В—А–∞–ґ–∞—О—Й–Є–є –њ–Њ—Б—В–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –љ–µ–Њ–±—А–∞—В–Є–Љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤ –Љ—Л—И—Ж–∞—Е –Є –і—А—Г–≥–Є—Е –Њ—А–≥–∞–љ–∞—Е (—А—Г–±—Ж–µ–≤–∞–љ–Є–µ, —Д–Є–±—А–Њ–Ј, –∞–Љ–Є–Њ—В—А–Њ—Д–Є–Є, –Ј–∞–Љ–µ—Й–µ–љ–Є–µ –ґ–Є—А–Њ–≤–Њ–є —В–Ї–∞–љ—М—О) –Є –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ –њ—А–Њ–≤–Њ–і–Є–Љ–Њ–є —В–µ—А–∞–њ–Є–Є (—Б—В–µ—А–Њ–Є–і–љ–∞—П –Ї–∞—В–∞—А–∞–Ї—В–∞, —Б–∞—Е–∞—А–љ—Л–є –і–Є–∞–±–µ—В, –Є–љ—Д–µ–Ї—Ж–Є–Њ–љ–љ—Л–µ –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є—П –Є –і—А.) [30–34]. –Я–Њ–ї–љ—Л–є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–є –Њ—В–≤–µ—В –љ–∞ —В–µ—А–∞–њ–Є—О –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П –Ї–∞–Ї –Њ—В—Б—Г—В—Б—В–≤–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –±–Њ–ї–µ–Ј–љ–Є –љ–∞ —Д–Њ–љ–µ –њ—А–Њ–і–Њ–ї–ґ–µ–љ–Є—П –њ—А–Њ–≤–Њ–і–Є–Љ–Њ–≥–Њ –ї–µ—З–µ–љ–Є—П, –∞ –њ–Њ–ї–љ–∞—П –Ї–ї–Є–љ–Є—З–µ—Б–Ї–∞—П —А–µ–Љ–Є—Б—Б–Є—П – –Ї–∞–Ї –Њ—В—Б—Г—В—Б—В–≤–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Љ–Є–Њ–Ј–Є—В–∞ –љ–∞ —Д–Њ–љ–µ –Њ—В—Б—Г—В—Б—В–≤–Є—П –ї—О–±–Њ–є —В–µ—А–∞–њ–Є–Є –≤ —В–µ—З–µ–љ–Є–µ 6 –Љ–µ—Б—П—Ж–µ–≤ [32].
–Ю—Б–љ–Њ–≤—Г –ї–µ—З–µ–љ–Є—П –Ш–Т–Ь —Б–Њ—Б—В–∞–≤–ї—П—О—В –У–Ъ [36]. –†–∞–љ–љ–µ–µ –љ–∞—З–∞–ї–Њ —В–µ—А–∞–њ–Є–Є (–≤ —В–µ—З–µ–љ–Є–µ –њ–µ—А–≤—Л—Е 3–—Е –Љ–µ—Б—П—Ж–µ–≤ –Њ—В –љ–∞—З–∞–ї–∞ —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤) –∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П —Б –±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–Љ –њ—А–Њ–≥–љ–Њ–Ј–Њ–Љ [37,38]. –Т –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В —В—П–ґ–µ—Б—В–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –љ–∞—З–∞–ї—М–љ–∞—П –і–Њ–Ј–∞ –Ї–Њ–ї–µ–±–ї–µ—В—Б—П –Њ—В 1 –і–Њ 2 –Љ–≥/–Ї–≥/—Б—Г—В. –£–ї—Г—З—И–µ–љ–Є–µ —Б–Њ—Б—В–Њ—П–љ–Є—П (–љ–∞—А–∞—Б—В–∞–љ–Є–µ –Љ—Л—И–µ—З–љ–Њ–є —Б–Є–ї—Л) –±–Њ–ї—М–љ—Л—Е –Я–Ь/–Ф–Ь —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –Љ–µ–і–ї–µ–љ–љ–µ–µ, —З–µ–Љ –њ—А–Є –і—А—Г–≥–Є—Е —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е. –Я—А–Є –Њ—В—Б—Г—В—Б—В–≤–Є–Є –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ–є –і–Є–љ–∞–Љ–Є–Ї–Є –≤ —В–µ—З–µ–љ–Є–µ 4 –љ–µ–і–µ–ї—М —Б–ї–µ–і—Г–µ—В —Г–≤–µ–ї–Є—З–Є—В—М –і–Њ–Ј—Г –У–Ъ. –Я–Њ—Б–ї–µ –і–Њ—Б—В–Є–ґ–µ–љ–Є—П —Н—Д—Д–µ–Ї—В–∞ –Є–ї–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–≥–Њ —Г–ї—Г—З—И–µ–љ–Є—П (–љ–∞—А–∞—Б—В–∞–љ–Є–µ –Љ—Л—И–µ—З–љ–Њ–є —Б–Є–ї—Л –Є –Ъ–§–Ъ) –і–Њ–Ј—Г –У–Ъ –њ–Њ—Б—В–µ–њ–µ–љ–љ–Њ —Б–љ–Є–ґ–∞—О—В –і–Њ –њ–Њ–і–і–µ—А–ґ–Є–≤–∞—О—Й–µ–є. –°–љ–Є–ґ–µ–љ–Є–µ –і–Њ–Ј—Л –У–Ъ –і–Њ–ї–ґ–љ–Њ –њ—А–Њ–≤–Њ–і–Є—В—М—Б—П –њ–Њ–і —Б—В—А–Њ–≥–Є–Љ –Ї–ї–Є–љ–Є–Ї–Њ––ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л–Љ –Ї–Њ–љ—В—А–Њ–ї–µ–Љ [39].
–Я—Г–ї—М—Б–—В–µ—А–∞–њ–Є—П –У–Ъ —А–µ–і–Ї–Њ —Н—Д—Д–µ–Ї—В–Є–≤–љ–∞, –њ—А–Є–Љ–µ–љ—П–µ—В—Б—П, –≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ, –њ—А–Є –Ѓ–Ф–Ь, –Ї–Њ–≥–і–∞ –Њ–љ–∞ –Љ–Њ–ґ–µ—В –њ—А–µ–і–Њ—В–≤—А–∞—В–Є—В—М –±—Л—Б—В—А–Њ–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –Љ–Є–Њ–њ–∞—В–Є–Є –Є —А–∞–Ј–≤–Є—В–Є–µ –Ї–∞–ї—М—Ж–Є–љ–Њ–Ј–∞. –Я—А–Є –Я–Ь/–Ф–Ь —Г –≤–Ј—А–Њ—Б–ї—Л—Е –њ—Г–ї—М—Б–—В–µ—А–∞–њ–Є—О –У–Ъ –њ—А–Є–Љ–µ–љ—П—О—В –≤ —Б–ї—Г—З–∞–µ –±—Л—Б—В—А–Њ–≥–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –і–Є—Б—Д–∞–≥–Є–Є (—А–Є—Б–Ї –∞—Б–њ–Є—А–∞—Ж–Є–Њ–љ–љ–Њ–є –њ–љ–µ–≤–Љ–Њ–љ–Є–Є) –Є —А–∞–Ј–≤–Є—В–Є—П —Б–Є—Б—В–µ–Љ–љ—Л—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є (–Љ–Є–Њ–Ї–∞—А–і–Є—В, –∞–ї—М–≤–µ–Њ–ї–Є—В).
–Я—А–Є –љ–∞–ї–Є—З–Є–Є —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞ –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ–Њ–≥–Њ –њ—А–Њ–≥–љ–Њ–Ј–∞ (–њ–Њ–Ј–і–љ–µ–µ –љ–∞–Ј–љ–∞—З–µ–љ–Є–µ –У–Ъ–—В–µ—А–∞–њ–Є–Є, —В—П–ґ–µ–ї–∞—П –Љ—Л—И–µ—З–љ–∞—П —Б–ї–∞–±–Њ—Б—В—М, –љ–∞–ї–Є—З–Є–µ –і–Є—Б—Д–∞–≥–Є–Є) [37,38], –њ—А–Є –љ–µ–≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –љ–∞–Ј–љ–∞—З–µ–љ–Є—П –∞–і–µ–Ї–≤–∞—В–љ–Њ–є –і–Њ–Ј—Л –У–Ъ –Є–Ј––Ј–∞ –њ–Њ–±–Њ—З–љ—Л—Е —Н—Д—Д–µ–Ї—В–Њ–≤ –Є–ї–Є –њ—А–Є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ–є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –У–Ъ –њ—А–Є–Љ–µ–љ—П—О—В—Б—П –њ—А–µ–њ–∞—А–∞—В—Л «–≤—В–Њ—А–Њ–≥–Њ» —А—П–і–∞: –Љ–µ—В–Њ—В—А–µ–Ї—Б–∞—В, –∞–Ј–∞—В–Є–Њ–њ—А–Є–љ [37], —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і: («–њ—А–µ–њ–∞—А–∞—В –≤—Л–±–Њ—А–∞» –њ—А–Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–Љ –ї–µ–≥–Њ—З–љ–Њ–Љ —Д–Є–±—А–Њ–Ј–µ), —Ж–Є–Ї–ї–Њ—Б–њ–Њ—А–Є–љ –Р.
–Ш–Љ–µ—О—В—Б—П –і–∞–љ–љ—Л–µ –Њ –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ–Љ —Н—Д—Д–µ–Ї—В–µ —Ж–Є–Ї–ї–Њ—Б–њ–Њ—А–Є–љ–∞ –Р –≤ –Њ—В–љ–Њ—И–µ–љ–Є–Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—П –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —Д–Є–±—А–Њ–Ј–∞ [40,41]. –Э–∞–Ї–Њ–њ–ї–µ–љ –љ–µ–Ї–Њ—В–Њ—А—Л–є –Њ–њ—Л—В –ї–µ—З–µ–љ–Є—П —В–∞–Ї—А–Њ–ї–Є–Љ—Г—Б–Њ–Љ, –Ї–Њ—В–Њ—А—Л–є —Б—Е–Њ–і–µ–љ –њ–Њ –Є–Љ–Љ—Г–љ–Њ–Љ–Њ–і—Г–ї–Є—А—Г—О—Й–Є–Љ —Н—Д—Д–µ–Ї—В–∞–Љ —Б —Ж–Є–Ї–ї–Њ—Б–њ–Њ—А–Є–љ–Њ–Љ –Р [39,42,43].
–Ю–і–љ–Є–Љ –Є–Ј –љ–∞–Є–±–Њ–ї–µ–µ –Њ–±–љ–∞–і–µ–ґ–Є–≤–∞—О—Й–Є—Е –љ–∞–њ—А–∞–≤–ї–µ–љ–Є–є —Д–∞—А–Љ–∞–Ї–Њ—В–µ—А–∞–њ–Є–Є –Ш–Т–Ь –Љ–љ–Њ–≥–Є–µ –≥–Њ–і—Л —Б—З–Є—В–∞–µ—В—Б—П –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –≤–љ—Г—В—А–Є–≤–µ–љ–љ–Њ–≥–Њ –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞. –Х—Й–µ –≤ 1993 –≥. –Ь.–°. Dalakas [44] –і–Њ–Ї–∞–Ј–∞–љ –µ–≥–Њ –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–є –Ї–ї–Є–љ–Є–Ї–Њ––ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л–є —Н—Д—Д–µ–Ї—В. –Ю–і–љ–∞–Ї–Њ –Ї—А–∞—В–Ї–Њ–≤—А–µ–Љ–µ–љ–љ–Њ—Б—В—М —Н—Д—Д–µ–Ї—В–∞ –њ–Њ—Б–ї–µ –њ—А–µ–Ї—А–∞—Й–µ–љ–Є—П –µ–≥–Њ –њ—А–Є–Љ–µ–љ–µ–љ–Є—П —В—А–µ–±—Г–µ—В –њ—А–Њ–≤–µ–і–µ–љ–Є—П –њ–Њ–≤—В–Њ—А–љ—Л—Е –Є–љ—Д—Г–Ј–Є–є. –Я–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ—Л–Љ –њ–Њ–Ї–∞–Ј–∞–љ–Є–µ–Љ –і–ї—П –≤–љ—Г—В—А–Є–≤–µ–љ–љ–Њ–≥–Њ –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ —П–≤–ї—П–µ—В—Б—П —В—П–ґ–µ–ї–∞—П –і–Є—Б—Д–∞–≥–Є—П [42].
–Ю—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ –љ–Њ–≤—Л–Љ —Ж–Є—В–Њ—Б—В–∞—В–Є—З–µ—Б–Ї–Є–Љ –њ—А–µ–њ–∞—А–∞—В–Њ–Љ –і–ї—П –ї–µ—З–µ–љ–Є—П –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є —П–≤–ї—П–µ—В—Б—П –Љ–Є–Ї–Њ—Д–µ–љ–Њ–ї–∞—В–∞ –Љ–Њ—Д–µ—В–Є–ї – –Є–Љ–Љ—Г–љ–Њ–і–µ–њ—А–µ—Б—Б–∞–љ—В –∞–љ—В–Є–Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Њ–≥–Њ —В–Є–њ–∞, –Њ–±–ї–∞–і–∞—О—Й–Є–є —Е–Њ—А–Њ—И–µ–є –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В—М—О —Б–Њ–≥–ї–∞—Б–љ–Њ –ї–Є—В–µ—А–∞—В—Г—А–љ—Л–Љ –і–∞–љ–љ—Л–Љ [42,43].
–Т –њ–Њ—Б–ї–µ–і–љ–Є–µ –≥–Њ–і—Л –і–ї—П –ї–µ—З–µ–љ–Є—П –Ш–Т–Ь –≤—Б–µ —З–∞—Й–µ –њ—А–Є–Љ–µ–љ—П—О—В—Б—П –Є–љ–≥–Є–±–Є—В–Њ—А—Л —Д–∞–Ї—В–Њ—А–∞ –љ–µ–Ї—А–Њ–Ј–∞ –Њ–њ—Г—Е–Њ–ї–Є? (–§–Э–Ю–a), –≤ —З–∞—Б—В–љ–Њ—Б—В–Є –Є–љ—Д–ї–Є–Ї—Б–Є–Љ–∞–± [42,45–48]. –Я–Њ—П–≤–Є–ї–Є—Б—М –і–∞–љ–љ—Л–µ –Њ–± —Г—Б–њ–µ—И–љ–Њ–Љ –њ—А–Є–Љ–µ–љ–µ–љ–Є–Є –њ—А–Є –∞—Г—В–Њ–Є–Љ–Љ—Г–љ–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤, –±–ї–Њ–Ї–Є—А—Г—О—Й–Є—Е –њ—А–Њ–ї–Є—Д–µ—А–∞—Ж–Є—О –Т––Ї–ї–µ—В–Њ–Ї, –Њ–і–љ–Є–Љ –Є–Ј –Ї–Њ—В–Њ—А—Л—Е —П–≤–ї—П–µ—В—Б—П —А–Є—В—Г–Ї—Б–Є–Љ–∞–±. –Ш–Љ–µ—О—В—Б—П –Њ—В–і–µ–ї—М–љ—Л–µ —Б–Њ–Њ–±—Й–µ–љ–Є—П –Њ–± –µ–≥–Њ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ—А–Є –Ф–Ь [42,49,50].
–Я—А–Є —А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ–Љ –Ї–Њ–ґ–љ–Њ–Љ —Б–Є–љ–і—А–Њ–Љ–µ, —Б–Њ—Е—А–∞–љ—П—О—Й–µ–Љ—Б—П –љ–∞ —Д–Њ–љ–µ –≤–Њ—Б—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –Љ—Л—И–µ—З–љ–Њ–є —Б–Є–ї—Л, —А–µ–Ї–Њ–Љ–µ–љ–і—Г–µ—В—Б—П –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –∞–љ—В–Є–Љ–∞–ї—П—А–Є–є–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤, —В–Њ–њ–Є—З–µ—Б–Ї–Є—Е —Б—В–µ—А–Њ–Є–і–Њ–≤, —В–∞–Ї—А–Њ–ї–Є–Љ—Г—Б–∞, –Љ–Є–Ї–Њ—Д–µ–љ–Њ–ї–∞—В –Љ–Њ—Д–µ—В–Є–ї–∞ [39,42,43].
–†–µ–∞–±–Є–ї–Є—В–∞—Ж–Є–Њ–љ–љ—Л–µ –Љ–µ—А–Њ–њ—А–Є—П—В–Є—П –њ—А–Њ–≤–Њ–і—П—В—Б—П –≤ –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В —Б—В–∞–і–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Т –Њ—Б—В—А—Г—О —Д–∞–Ј—Г –њ–Њ–Ї–∞–Ј–∞–љ—Л –њ–∞—Б—Б–Є–≤–љ—Л–µ —Г–њ—А–∞–ґ–љ–µ–љ–Є—П, –≤ —Б—В–∞–і–Є—О –≤—Л–Ј–і–Њ—А–Њ–≤–ї–µ–љ–Є—П – –Є–Ј–Њ–Љ–µ—В—А–Є—З–µ—Б–Ї–Є–µ, –∞ –Ј–∞—В–µ–Љ –Є–Ј–Њ—В–Њ–љ–Є—З–µ—Б–Ї–Є–µ —Г–њ—А–∞–ґ–љ–µ–љ–Є—П –Є, –љ–∞–Ї–Њ–љ–µ—Ж, –≤ —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є —Б—В–∞–і–Є–Є – –∞–љ–∞—Н—А–Њ–±–љ—Л–µ —Г–њ—А–∞–ґ–љ–µ–љ–Є—П [1].
–Я—А–Њ–≥–љ–Њ–Ј. –Т–љ–µ–і—А–µ–љ–Є–µ –≤ –Ї–ї–Є–љ–Є—З–µ—Б–Ї—Г—О –њ—А–∞–Ї—В–Є–Ї—Г –У–Ъ —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —Г–≤–µ–ї–Є—З–Є–ї–Њ –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М –±–Њ–ї—М–љ—Л—Е –Я–Ь/–Ф–Ь, –Ї–Њ—В–Њ—А–∞—П —Б–Њ—Б—В–∞–≤–ї—П–µ—В 90% —З–µ—А–µ–Ј 5 –ї–µ—В –њ–Њ—Б–ї–µ –њ–Њ—Б—В–∞–љ–Њ–≤–Ї–Є –і–Є–∞–≥–љ–Њ–Ј–∞, –Ј–∞ –Є—Б–Ї–ї—О—З–µ–љ–Є–µ–Љ –±–Њ–ї—М–љ—Л—Е –Њ–љ–Ї–Њ–Љ–Є–Њ–Ј–Є—В–Њ–Љ. –§–∞–Ї—В–Њ—А–∞–Љ–Є, –∞—Б—Б–Њ—Ж–Є–Є—А—Г—О—Й–Є–Љ–Є—Б—П —Б –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–Љ –њ—А–Њ–≥–љ–Њ–Ј–Њ–Љ –њ—А–Є –Я–Ь/–Ф–Ь, —П–≤–ї—П—О—В—Б—П –њ–Њ–ґ–Є–ї–Њ–є –≤–Њ–Ј—А–∞—Б—В, –њ–Њ–Ј–і–љ–Њ –њ–Њ—Б—В–∞–≤–ї–µ–љ–љ—Л–є –і–Є–∞–≥–љ–Њ–Ј, –љ–µ–∞–і–µ–Ї–≤–∞—В–љ–∞—П —В–µ—А–∞–њ–Є—П –У–Ъ –≤ –љ–∞—З–∞–ї–µ –±–Њ–ї–µ–Ј–љ–Є, —В—П–ґ–µ–ї–Њ–µ —В–µ—З–µ–љ–Є–µ –Љ–Є–Њ–Ј–Є—В–∞, –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–є —Б–Є–љ–і—А–Њ–Љ [1,8].






–Ы–Є—В–µ—А–∞—В—Г—А–∞
1. –Э–∞—Б–Њ–љ–Њ–≤ –Х.–Ы. –Т–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Љ—Л—И—Ж. –†–∞—Ж–Є–Њ–љ–∞–ї—М–љ–∞—П —Д–∞—А–Љ–∞–Ї–Њ—В–µ—А–∞–њ–Є—П —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є. 195–202. –Ь–Њ—Б–Ї–≤–∞. –Ы–Є—В–µ—А–∞. 2003.
2. Wortmann R.L. “Diseases of Skeletal Muscle” 1 —Б 45–57.
3. Dalakas M. Mechanisms of disease:signaling pathways and immunobyology of inflammatory myopathies. Nature clinical practice rheumatology. 2006, vol l2, 4, 219–227.
4. –Э–∞—Б–Њ–љ–Њ–≤ –Х.–Ы., –®—В—Г—В–Љ–∞–љ –Т.–Ч, –°–∞–ї–Њ–ґ–Є–љ –Ъ.–Т, –У—Г—Б–µ–≤–∞ –Э.–У., –Э–∞—Б–Њ–љ–Њ–≤–∞ –Т.–Р, –Я–ї–Њ—В—Ж –Я. –Ъ–ї–Є–љ–Є–Ї–Њ – –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –≥–µ—В–µ—А–Њ–≥–µ–љ–љ–Њ—Б—В—М –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є—Е –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Љ–Є–Њ–њ–∞—В–Є–є. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ь–µ–і–Є—Ж–Є–љ–∞ 1995; 2 3–8.
5. Miller M. Clinical manifestations and diagnosis of adult dermatomyositis and po;ymyositis. UpToDate 2004; 12.2
6. Stahl, NI, Klippel, JH, Decker, JL. A cutaneous lesion associated with myositis. Ann Intern Med 1979; 91:577
7. Kovacs, SO, Kovacs, SC. Dermatomyositis. J Am Acad Dermatol 1998; 39:899.
8. –С–Њ–љ–і–∞—А–µ–љ–Ї–Њ –Ш.–Т., –Ь—Г—Е–Є–љ –Э.–Р., –Э–∞—Б–Њ–љ–Њ–≤ –Х.–Ы. –Я–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е –њ—А–Є –њ–Њ–ї–Є–Љ–Є–Њ–Ј–Є—В–µ –Є –і–µ—А–Љ–∞—В–Њ–Љ–Є–Њ–Ј–Є—В–µ. –Ш–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –ї–µ–≥–Ї–Є—Е. –†—Г–Ї–Њ–≤–Њ–і—Б—В–≤–Њ –і–ї—П –≤—А–∞—З–µ–є. –Я–Њ–і —А–µ–і–∞–Ї—Ж–Є–µ–є –Ш–ї—М–Ї–Њ–≤–Є—З–∞ –Ь.–Ь., –Ъ–Њ–Ї–Њ—Б–Њ–≤–∞ –Р.–Э. 274–287. –°–∞–љ–Ї—В––Я–µ—В–µ—А—Г—А–≥. –Э–Њ—А–і–Љ–µ–і–Є–Ј–і–∞—В 2005.
9. Lakhanpal, S, Lie, JT, Conn, DL, Martin, WJ II. Pulmonary disease in polymyositis/dermatomyositis: A clinicopathologic analysis of 65 autopsied cases. Ann Rheum Dis 1987; 46:23.
10. –•–Є—В—А–Њ–≤ –Р.–Э. –Я–Њ—А–∞–ґ–µ–љ–Є–µ —Б–µ—А–і—Ж–∞ –њ—А–Є –і–µ—А–Љ–∞—В–Њ–Љ–Є–Њ–Ј–Є—В–µ –Є –њ–Њ–ї–Є–Љ–Є–Њ–Ј–Є—В–µ. –Р–≤—В–Њ—А–µ—Д–µ—А–∞—В –і–Є—Б—Б–µ—А—В–∞—Ж–Є–Є –љ–∞ —Б–Њ–Є—Б–Ї–∞–љ–Є–µ —Г—З–µ–љ–Њ–є —Б—В–µ–њ–µ–љ–Є –Ї–∞–љ–і–Є–і–∞—В–∞ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Є—Е –љ–∞—Г–Ї. –Ь–Њ—Б–Ї–≤–∞ 1999.
11. Love, LA, Leff, RL, Fraser, DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: Myositisspecific autoantibodies define useful homogeneous patient groups. Medicine 1991; 70:360.
12. Bohan, A, Peter, JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975; 292:403.
13. Bohan, A, Peter, JB, Bowman, RL, Pearson, CM. Computer–assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255.
14. Reichlin, M, Arnett, FC. Multiplicity of antibodies in myositis sera. Arthritis Rheum 1984; 27:1150.
15. Love, LA, Leff, RL, Fraser, DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: Myositisspecific autoantibodies define useful homogeneous patient groups. Medicine 1991; 70:360.
16. Plotz, PH, Rider, LG, Targoff, IN, et al. Myositis: Immunologic contributions to understanding cause, pathogenesis, and therapy. Ann Intern Med 1995; 122:715.
17. Brouwer, R, Hengstman, GJ, Vree Egberts, W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann. Rheum Dis 2001; 60:116.
18. Mozaffar T, Pestronk, A. Myopathy with anti–Jo–1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472.
19. –У–µ—Е—В –С.–Ь., –Ш–ї—М–Є–љ–∞ –Э.–Р. «–Э–µ—А–≤–љ–Њ––Љ—Л—И–µ—З–љ—Л–µ –±–Њ–ї–µ–Ј–љ–Є». «–Ь–µ–і–Є—Ж–Є–љ–∞» 1982.
20. –Р–љ—В–µ–ї–∞–≤–∞ –Ю.–Р., –Ъ–∞—Б–∞—В–Ї–Є–љ–∞ –Ы.–§., –У—Г—А–Ї–Є–љ–∞ –У.–Ґ., –•–Є—В—А–Њ–≤ –Р.–Э., –Я–Є–Ї—Г–ї—П –Э.–Т., –®—В—Г—В–Љ–∞–љ –Т.–Ч., –Э–∞—Б–Њ–љ–Њ–≤ –Х.–Ы.. –Ф–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ–∞—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞ –Љ—Л—И–µ—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В–Є. –†—Г—Б—Б–Ї–Є–є –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Є–є –ґ—Г—А–љ–∞–ї N 14, 2004, 854–862.
21. Dalakas M.C. Polymyositis, dermatomyositis and inclusion–body myositis. Engl.J. Med. 1991, 325, 1487–1498.
22. Miller FW, Rider LG, Chung YL, et al, for the International Myositis Outcome Assessment Collaborative Study Group, Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies, Rheumatology (Oxford), 2001, 40,11,1262–1273.).
23. Griggs RC, Askanas, V, DiMauro, S, et al. Inclusion body myositis and myopathies. Ann Neurol 1995, 38:705.
24. Tanimoto K., Nakano K., Kano S. et al. Classification criteria for polymyositis and dermatomyositis. J. Rheumatol 1995; 22: 668–574.
25. Targoff I.N., Miller F.W., Medsger T.A. et al. Classification criteria for the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 1997;9, 527–535.
26. Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies: epidemiology, classification, and diagnostic criteria.Rheum Dis Clin North Am 2002;28, 823–832.
27. Dalakas M.C., Hohlfeld R. Pofymyositis and dermatomyositis. Lancet 2003, 362, 971–982.
28. Nirmalananthan N, Holton JL. Hanna MG. Is it really myositis? Consideration of the differential diagnosis. Curr Opin Rheumatol 2004; 16: 684–691.
29. –Ш–ї—М–Є–љ–∞ –Э.–Ш. «–Ь–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–µ —Б–Є–љ–і—А–Њ–Љ—Л» –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Љ–µ–і–Є—Ж–Є–љ–∞ , 1983, N9, —Б 30–35.
30. Isenberg D.A., Allen E., Farewell V., et al. International consensus outcome measures for patients with idiopathic inflammatory myopathies. Development and initial validation of myositis activity and damage indices in patients with adult onset disease. Rheumatology (Oxford), 2004, 43, 1, 49–54.
31. Miller F.W., Rider L.G., Feldman. Development of validated disease activity and damage indices for the juvenile idiopathic inflammatory myopathies: I. Arthr. Rheum., 1997, 40, 11, 1976–1983.
32. Oddis C.V., Outcomes and disease activity measures for assessing treatments in the idiopathic inflammatory myopathies. Curr. Rheumatol. Rep. 2005, 7, 87–93.
33. Lovell D.J., Lindsley C.B., Rennebohm R.M., et al. Development of validated disease activity and damage indices for the juvenile idiopathic inflammatory myopathies. II. The Childhood Myositis Assessment Scale (CMAS): a quantitative tool for the evaluation of muscle function. The Juvenile Dermatomyositis Disease Activity Collaborative Study Group. Arthritis Rheum. 1999 42, 10, 2213–2219.
34. Park J. H., Olsen N. J, King L. et al. Use of imaging and P–31 magnetic resonance spectroscopy to detect and quantify muscle dysfunction in the myopathic variants of dermatomyositis. Arth.Rheum., 1995, 38, 68–77.
35. Lovell D.J., Lindsley C.B., Rennebohm R.M., et al. Development of validated disease activity and damage indices for the juvenile idiopathic inflammatory myopathies. II. The Childhood Myositis Assessment Scale (CMAS): a quantitative tool for the evaluation of muscle function. The Juvenile Dermatomyositis Disease Activity Collaborative Study Group. Arthritis Rheum. 1999 42, 10, 2213–2219.
36. Drake LA, Dinehart SM, Farmer ER, et al. Guidelines of care for dermatomyositis. J Am Acad Dermatol 1996; 34:824.
37. Fafalak RG, Peterson MGE, Kagen LJ. Strength in polymyositis and dermatomyositis: Best outcome in patients treated early. J Rheumatol. 1994; 21:643.
38. Joffe MM, Love LA, Leff RL et al. Drug therapy of the idiopathic inflammatory myopathies: Predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379.
39. –Э–∞—Б–Њ–љ–Њ–≤ –Х.–Ы., –®—В—Г—В–Љ–∞–љ –Т.–Ч. –§–∞—А–Љ–∞–Ї–Њ—В–µ—А–∞–њ–Є—П –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є—Е –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Љ–Є–Њ–њ–∞—В–Є–є. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П —Д–∞—А–Љ–∞–Ї–Њ–ї–Њ–≥–Є—П, 1995, 4 (2). 57–63.
40. Grau JM, Herrero, C, Casademont J, et al. Cyclosporin A as a first choice therapy for dermatomyositis. J Rheumatol 1994; 21:381.
41. Qushmaq KA, Chalmers A, Esdaile JM. Cyclosporin A in the treatment of refractory adult polymyositis/dermatomyositis: population based experience in 6 patients and literature review. J Rheumatol 2000; 27:2855.
42. –Р–љ—В–µ–ї–∞–≤–∞ –Ю.–Р., –°–Њ–ї–Њ–≤—М–µ–≤ –°.–Ъ,. –•–Є—В—А–Њ–≤ –Р.–Э, –Э–∞—Б–Њ–љ–Њ–≤ –Х.–Ы. –Э–Њ–≤—Л–µ –∞—Б–њ–µ–Ї—В—Л —Д–∞—А–Љ–∞–Ї–Њ—В–µ—А–∞–њ–Є–Є –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є—Е –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Љ–Є–Њ–њ–∞—В–Є–є. (–Ю–±–Ј–Њ—А –ї–Є—В–µ—А–∞—В—Г—А—Л). –†—Г—Б—Б–Ї–Є–є –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Є–є –ґ—Г—А–љ–∞–ї 2006, 14, 8 (260), 627–629.
43. –Р–љ—В–µ–ї–∞–≤–∞ –Ю.–Р., –Э–∞—Б–Њ–љ–Њ–≤ –Х.–Ы. –Ь–µ—Б—В–Њ –Љ–Є–Ї–Њ—Д–µ–љ–Њ–ї–∞—В –Љ–Њ—Д–µ—В–Є–ї–∞ –њ—А–Є –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є—Е –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Љ–Є–Њ–њ–∞—В–Є—П—Е. –Э–∞—Г—З–љ–Њ––њ—А–∞–Ї—В–Є—З–µ—Б–Ї–∞—П —А–µ–≤–Љ–∞—В–Њ–ї–Њ–≥–Є—П 2006,3,38–41.
44. Dalakas, MC, Illa I, Dambrosia, JM, et al. A controlled trial of high–dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 1993; 329:1993
45. Efthimiou P., Schwartzman S, Kagen L. Possible role for TNF–Inhibitors in the treatment of resistant dermatomyositis and polymiositis. Ann.Rheum. Dis. 2006, 13.
46. Hengstelman G.J., van den Hoogen F.H., Barrera P. et –∞l. Successful treatment of dermatomyositis and polymyositis with anti–tumor–necrosis–factor–alpha: preliminary observations. Eur.Neurol., 2003, 50(1),10–15.
47. Saaden C.K. Etanercept is effective in the treatment of polymyositis/dermatomyositis wich refractory to conventional therapy including steroid and other disease modifying agents. Arthr.Rheum., 2000, 43,193.
48. Hengstelman G., van den Hoogen F., van Engelen B., et al. Anti–TNF–blockade with infliximab (Remicaid) in polymyositis and dermatomyositis. Arthr.Rheum., 2000,43, 3193.
49. Levin T.D. Rituximab in the treatment of dermatomyositis. Arthr. Rheum. 2005, 52, 2, 601–607.
50. Levin T.D. A pilot study of rituximab therapy for refractory dermatomyositis. Arthr. Rheum. 2002,46, (suppl 9), 488.



