




–Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –≤—Б—В—А–µ—З–∞—О—Й–Є–Љ—Б—П –њ—А–Є –Я–Ь/–Ф–Ь (45–50%) —П–≤–ї—П–µ—В—Б—П –њ–Њ—А–∞–ґ–µ–љ–Є–µ –і—Л—Е–∞—В–µ–ї—М–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л [8,9]. –†–∞–Ј–≤–Є–≤–∞—О—Й–Є–µ—Б—П –њ—А–Є —Н—В–Њ–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П —Б–Њ —Б—В–Њ—А–Њ–љ—Л –Њ—А–≥–∞–љ–Њ–≤ –і—Л—Е–∞–љ–Є—П –≤–µ—Б—М–Љ–∞ —А–∞–Ј–љ–Њ–Њ–±—А–∞–Ј–љ—Л –Є –њ—А–Є–≤–ї–µ–Ї–∞—О—В –≤–љ–Є–Љ–∞–љ–Є–µ –Ї–∞–Ї —А–µ–≤–Љ–∞—В–Њ–ї–Њ–≥–Њ–≤, —В–∞–Ї –Є –њ—Г–ї—М–Љ–Њ–љ–Њ–ї–Њ–≥–Њ–≤, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Њ–љ–Є –Є–љ–Њ–≥–і–∞ –Њ–њ–µ—А–µ–ґ–∞—О—В –Ї–∞—А—В–Є–љ—Г –Љ—Л—И–µ—З–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤.
–Я–µ—А–≤—Л–µ —А–∞–±–Њ—В—Л –њ–Њ –њ—А–Њ–±–ї–µ–Љ–µ –ї–µ–≥–Њ—З–љ—Л—Е –њ–Њ—А–∞–ґ–µ–љ–Є–є –њ—А–Є –Я–Ь/–Ф–Ь –Њ—В–љ–Њ—Б—П—В—Б—П –Ї 1940 –≥., –Ї–Њ–≥–і–∞ P.A. O’Leary, M. Waisman [10] –Њ–њ–Є—Б–∞–ї–Є —Б–ї—Г—З–∞–Є –∞—Б–њ–Є—А–∞—Ж–Є–Њ–љ–љ–Њ–є –њ–љ–µ–≤–Љ–Њ–љ–Є–Є —Г –±–Њ–ї—М–љ—Л—Е –Ф–Ь. –Т –і–∞–ї—М–љ–µ–є—И–µ–Љ –±—Л–ї–Є –Њ–њ–Є—Б–∞–љ—Л –Є –і—А—Г–≥–Є–µ –≤–∞—А–Є–∞–љ—В—Л –њ–Њ—А–∞–ґ–µ–љ–Є—П –і—Л—Е–∞—В–µ–ї—М–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –њ—А–Є –Я–Ь/–Ф–Ь, –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–ґ–љ–Њ –Њ–±—К–µ–і–Є–љ–Є—В—М –≤ –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ –≥—А—Г–њ–њ, –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л—Е –≤ —В–∞–±–ї–Є—Ж–µ 1.
–Я—А–Є –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –±–Є–Њ–њ—В–∞—В–Њ–≤ –ї–µ–≥–Ї–Є—Е –±–Њ–ї—М–љ—Л—Е —Б —Б–Є–љ–і—А–Њ–Љ–Њ–Љ —Д–Є–±—А–Њ–Ј–Є—А—Г—О—Й–µ–≥–Њ –∞–ї—М–≤–µ–Њ–ї–Є—В–∞ (–°–§–Р) –Є –Я–Ь/–Ф–Ь –≤—Л—П–≤–ї—П–µ—В—Б—П –љ–µ—А–∞–≤–љ–Њ–Љ–µ—А–љ–Њ–µ —А–∞—Б–њ—А–µ–і–µ–ї–µ–љ–Є–µ –њ–Њ—А–∞–ґ–µ–љ–љ—Л—Е —Г—З–∞—Б—В–Ї–Њ–≤ –Љ–µ–ґ–і—Г —Г—З–∞—Б—В–Ї–∞–Љ–Є –Ј–і–Њ—А–Њ–≤–Њ–є —В–Ї–∞–љ–Є. –Т—Л—П–≤–ї–µ–љ–∞ –≥–Є–њ–µ—А–њ–ї–∞–Ј–Є—П –њ–љ–µ–≤–Љ–Њ—Ж–Є—В–Њ–≤ 2 —В–Є–њ–∞, —Г—В–Њ–ї—Й–µ–љ–Є–µ –∞–ї—М–≤–µ–Њ–ї—П—А–љ—Л—Е —Б—В–µ–љ–Њ–Ї –Є –њ–µ—А–µ–≥–Њ—А–Њ–і–Њ–Ї, —А–∞–Ј—А–∞—Б—В–∞–љ–Є–µ –њ–µ—А–Є–±—А–Њ–љ—Е–Є–∞–ї—М–љ–Њ–є –Є –њ–µ—А–Є–≤–∞—Б–Ї—Г–ї—П—А–љ–Њ–є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–∞–Ї–љ–Є, –∞ —В–∞–Ї–ґ–µ, –≥–Є–њ–µ—А–њ–ї–∞—Б—В–Є—З–µ—Б–Ї–Є–є —Б–Ї–ї–µ—А–Њ–Ј –Љ–∞–ї—Л—Е –∞—А—В–µ—А–Є–є. –Т–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–є –Є–љ—Д–Є–ї—М—В—А–∞—В —Б–Њ—Б—В–Њ–Є—В –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –Є–Ј –ї–Є–Љ—Д–Њ—Ж–Є—В–Њ–≤ –Є –њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї; —Н–Њ–Ј–Є–љ–Њ—Д–Є–ї—Л –Є –ї–µ–є–Ї–Њ—Ж–Є—В—Л –≤—Б—В—А–µ—З–∞—О—В—Б—П —А–µ–і–Ї–Њ [9].
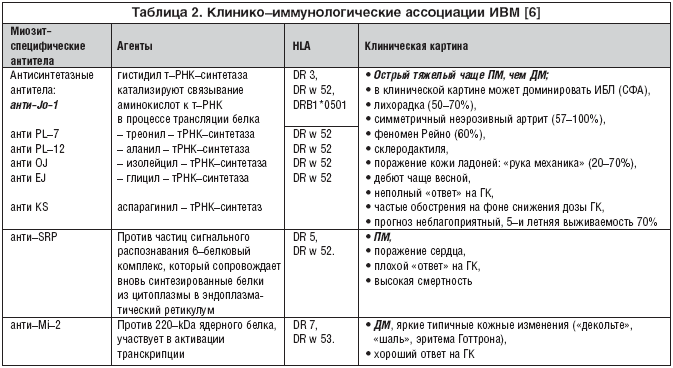
–Ъ–ї–Є–љ–Є–Ї–Њ––Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –∞—Б—Б–Њ—Ж–Є–∞—Ж–Є—П. –Р–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–є —Б–Є–љ–і—А–Њ–Љ. –Т 80–—Е –≥–Њ–і–∞—Е –•–• –≤–µ–Ї–∞ –Њ—В–Љ–µ—З–µ–љ–Њ –Є –і–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –њ—А–Є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Љ–Є–Њ–њ–∞—В–Є—П—Е –љ–∞–±–ї—О–і–∞—О—В—Б—П –∞—Б—Б–Њ—Ж–Є–∞—Ж–Є–Є –Њ–њ—А–µ–і–µ–ї–µ–љ–љ–Њ–≥–Њ —Б–Є–Љ–њ—В–Њ–Љ–Њ–Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ —Б —А—П–і–Њ–Љ –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Љ–∞—А–Ї–µ—А–Њ–≤ (–Љ–Є–Њ–Ј–Є—В–—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–Љ–Є –∞–љ—В–Є—В–µ–ї–∞–Љ–Є – –Ь–°–Р). –Ґ–∞–Ї, —А–∞–Ј–≤–Є—В–Є–µ –Я–Ь/–Ф–Ь —Б–≤—П–Ј–∞–љ–Њ —Б —Б–Є–љ—В–µ–Ј–Њ–Љ —Ж–µ–ї–Њ–≥–Њ «—Б–µ–Љ–µ–є—Б—В–≤–∞» –∞—Г—В–Њ–∞–љ—В–Є—В–µ–ї, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л—Е –Ї —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є–Љ –±–µ–ї–Ї–∞–Љ –Є —А–Є–±–Њ–љ—Г–Ї–ї–µ–Є–љ–Њ–≤—Л–Љ –Ї–Є—Б–ї–Њ—В–∞–Љ. –Ь–°–Р –Њ–њ—А–µ–і–µ–ї—П—О—В—Б—П —Г 50% –±–Њ–ї—М–љ—Л—Е –Я–Ь/–Ф–Ь [6].
–Я–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –≤—Л—П–≤–ї–µ–љ–Є–µ –Ь–°–Р –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–Њ —Б –Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л–Љ–Є –Ї–ї–Є–љ–Є–Ї–Њ––Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є –њ–Њ–і–≥—А—Г–њ–њ–∞–Љ–Є. –Ґ–∞–Ї, –∞–љ—В–Є–SRP (–∞–љ—В–Є—В–µ–ї–∞, –Ї–Њ—В–Њ—А—Л–µ —А–µ–∞–≥–Є—А—Г–µ—В —Б —З–∞—Б—В–Є—Ж–∞–Љ–Є —Б–Є–≥–љ–∞–ї—М–љ–Њ–≥–Њ —А–∞—Б–њ–Њ–Ј–љ–∞–≤–∞–љ–Є—П, –њ—А–µ–і—Б—В–∞–≤–ї—П—О—Й–Є–Љ–Є –Ї–Њ–Љ–њ–ї–µ–Ї—Б –Є–Ј 6––Є –±–µ–ї–Ї–Њ–≤ –Є –Љ–Њ–ї–µ–Ї—Г–ї—Л –†–Э–Ъ, –Њ–±–µ—Б–њ–µ—З–Є–≤–∞—О—Й–Є–Љ–Є –њ–µ—А–µ–љ–Њ—Б –≤–љ–Њ–≤—М —Б–Є–љ—В–µ–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е –Љ–Њ–ї–µ–Ї—Г–ї –Ї —Н–љ–і–Њ–њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Њ–Љ—Г —А–µ—В–Є–Ї—Г–ї—Г–Љ—Г) –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л —Г –±–Њ–ї—М–љ—Л—Е —Б —В—П–ґ–µ–ї–Њ–є –њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–є –Љ–Є–Њ–њ–∞—В–Є–µ–є, –і–Є—Б—Д–∞–≥–Є–µ–є –Є –њ–ї–Њ—Е–Є–Љ «–Њ—В–≤–µ—В–Њ–Љ» –љ–∞ —В–µ—А–∞–њ–Є—О –У–Ъ. –Р–љ—В–Є––Ьi–2 (–±–µ–ї–Ї–Њ–≤–Њ–—П–і–µ—А–љ—Л–є –Ї–Њ–Љ–њ–ї–µ–Ї—Б) – –≤—Л—П–≤–ї—П—О—В—Б—П —Г –±–Њ–ї—М–љ—Л—Е –Ф–Ь —Б –Ї–Њ–ґ–љ—Л–Љ–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є, —Е–Њ—А–Њ—И–Є–Љ «–Њ—В–≤–µ—В–Њ–Љ» –љ–∞ –ї–µ—З–µ–љ–Є–µ –У–Ъ –Є –њ—А–Њ–≥–љ–Њ–Ј–Њ–Љ [6]. –Ю—Б–љ–Њ–≤–љ—Л–µ –Ї–ї–Є–љ–Є–Ї–Њ––Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –∞—Б—Б–Њ—Ж–Є–∞—Ж–Є–Є –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ—Л –≤ —В–∞–±–ї–Є—Ж–µ 2.
–Э–µ–і–∞–≤–љ–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ –њ–Њ–і—В–Є–њ –±–Њ–ї—М–љ—Л—Е —Б –∞–Љ–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–Љ –Ф–Ь –Є –∞–љ—В–Є—В–µ–ї–∞–Љ–Є –Ї –њ–Њ–ї–Є–њ–µ–њ—В–Є–і—Г 140 kDa –±—Л–ї –Њ–њ–Є—Б–∞–љ –≤ –∞—Б—Б–Њ—Ж–Є–∞—Ж–Є–Є —Б –Ш–С–Ы [12].
–Э–∞–Є–±–Њ–ї–µ–µ –Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ–∞—П –≥—А—Г–њ–њ–∞ –Ь–°–Р – —Н—В–Њ –≥—А—Г–њ–њ–∞ –±–Њ–ї—М–љ—Л—Е —Б –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–Љ–Є –∞–љ—В–Є—В–µ–ї–∞–Љ–Є (–∞–љ—В–Є—В–µ–ї–∞–Љ–Є –Ї –∞–Љ–Є–љ–Њ–∞—Ж–Є–ї––†–Э–Ъ–—Б–Є–љ—В–µ—В–∞–Ј–∞–Љ), —Д—Г–љ–Ї—Ж–Є—П –Ї–Њ—В–Њ—А—Л—Е –Ј–∞–Ї–ї—О—З–∞–µ—В—Б—П –≤ –Ї–∞—В–∞–ї–Є–Ј–µ –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ —Б–≤—П–Ј—Л–≤–∞–љ–Є—П –Њ—В–і–µ–ї—М–љ—Л—Е –∞–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В —Б —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–µ–є —В––†–Э–Ъ. –Э–∞–Є–±–Њ–ї–µ–µ –Є–Ј—Г—З–µ–љ–љ—Л–µ –Є–Ј –љ–Є—Е – –∞–љ—В–Є J–Њ–1 –∞–љ—В–Є—В–µ–ї–∞. –Ю–і–љ–∞–Ї–Њ —Б—Г—Й–µ—Б—В–≤—Г—О—В –Є –і—А—Г–≥–Є–µ –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–µ –∞–љ—В–Є—В–µ–ї–∞ (PL–7, PL–12, KS, OJ, EJ), –Ї–Њ—В–Њ—А—Л–µ –≤—Б—В—А–µ—З–∞—О—В—Б—П —А–µ–ґ–µ. –Ш—Е –љ–∞–ї–Є—З–Є–µ, —В–∞–Ї–ґ–µ —Б–Њ–њ—А—П–ґ–µ–љ–Њ —Б —Б–Є–Љ—В–Њ–Љ–Њ–Ї–Њ–Љ–њ–ї–µ–Ї—Б–Њ–Љ, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–Љ –і–ї—П –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞, –≤ —В.—З. —Б –Ш–С–Ы. –І–∞—Б—В–Њ—В–∞ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–є –њ–љ–µ–≤–Љ–Њ–љ–Є–Є —Г –±–Њ–ї—М–љ—Л—Е —Б –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 60–80%. –Ш–љ—В–µ—А–µ—Б–љ–Њ, —З—В–Њ –Њ–њ–Є—Б–∞–љ–Њ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е –≤ –Њ—В—Б—Г—В—Б—В–≤–Є–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є —П–≤–љ–Њ–≥–Њ –Љ–Є–Њ–Ј–Є—В–∞ —Г –±–Њ–ї—М–љ—Л—Е —Б –∞–љ—В–Є—В–µ–ї–∞–Љ–Є PL–12, KS, OJ [12].
–Р–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–є —Б–Є–љ–і—А–Њ–Љ —П–≤–ї—П–µ—В—Б—П –љ–∞–Є–±–Њ–ї–µ–µ —В—П–ґ–µ–ї—Л–Љ, —З—В–Њ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ, –≤ –њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М, –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є –°–§–Р. –°–Є–Љ–њ—В–Њ–Љ–Њ–Ї–Њ–Љ–њ–ї–µ–Ї—Б –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П —Б–ї–µ–і—Г—О—Й–Є–Љ–Є –Њ—Б–љ–Њ–≤–љ—Л–Љ–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ–Є –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є: –Њ—Б—В—А–Њ–µ –љ–∞—З–∞–ї–Њ, –ї–Є—Е–Њ—А–∞–і–Ї–∞, –Љ–Є–Њ–Ј–Є—В, —Б–Є–Љ–Љ–µ—В—А–Є—З–љ—Л–є –љ–µ—Н—А–Њ–Ј–Є–≤–љ—Л–є –∞—А—В—А–Є—В –Є–ї–Є –∞—А—В—А–∞–ї–≥–Є–Є, —Д–µ–љ–Њ–Љ–µ–љ –†–µ–є–љ–Њ, –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Ї–Њ–ґ–Є –ї–∞–і–Њ–љ–µ–є –њ–Њ —В–Є–њ—Г «—А—Г–Ї–Є –Љ–µ—Е–∞–љ–Є–Ї–∞» (—А–Є—Б. 1), –і–Є—Б—Д–∞–≥–Є—П, —Б–Ї–ї–µ—А–Њ–і–∞–Ї—В–Є–ї–Є—П –≤ –∞—Б—Б–Њ—Ж–Є–∞—Ж–Є–Є —Б –≤—Л—П–≤–ї–µ–љ–Є–µ–Љ –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л—Е –∞–љ—В–Є—В–µ–ї (Jo–1, —А–µ–ґ–µ –і—А—Г–≥–Є—Е). –Я—А–Є–Љ–µ—З–∞—В–µ–ї—М–љ–Њ, —З—В–Њ –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ–Њ–Љ—Г —Б–Є–љ–і—А–Њ–Љ—Г —Е–∞—А–∞–Ї—В–µ—А–љ–∞ —Б–µ–Ј–Њ–љ–љ–Њ—Б—В—М – –і–µ–±—О—В –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –≤ –≤–µ—Б–µ–љ–љ–Є–є –њ–µ—А–Є–Њ–і. –Т –њ–Њ—Б–ї–µ–і–љ–Є–µ –њ—А–Њ–≤–µ–і–µ–љ —А—П–і –Ї–ї–Є–љ–Є–Ї–Њ––Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ—Л—Е –љ–Є–ґ–µ, –Њ—В–Ї—А—Л–≤—И–Є—Е –љ–Њ–≤—Л–µ –≤–Ј–≥–ї—П–і—Л –љ–∞ —В–µ—З–µ–љ–Є–µ –Є –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞.
–Т 2006 Yoshifuji —Б —Б–Њ–∞–≤—В. [13] –њ—А–Њ–≤–µ–і–µ–љ–Њ –Є–љ—В–µ—А–µ—Б–љ–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ, –њ–Њ–Ї–∞–Ј–∞–≤—И–µ–µ –≤—Л—Б–Њ–Ї—Г—О —З–∞—Б—В–Њ—В—Г (–і–Њ 95%) –≤—Б—В—А–µ—З–∞–µ–Љ–Њ—Б—В–Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–є –њ–љ–µ–≤–Љ–Њ–љ–Є–Є —Г –±–Њ–ї—М–љ—Л—Е —Б –Ь–°–Р. –£ –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –і–µ–±—О—В–Є—А–Њ–≤–∞–≤—И–Є—Е —Б –ї–µ–≥–Њ—З–љ–Њ–є —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–Є –і–Њ —А–∞–Ј–≤–Є—В–Є—П –Љ—Л—И–µ—З–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞, —З–∞—Б—В–Њ—В–∞ –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л—Е –∞–љ—В–Є—В–µ–ї —Б–Њ—Б—В–∞–≤–ї—П–ї–∞ 75%. –Т —В–Њ –ґ–µ –≤—А–µ–Љ—П —Г –±–Њ–ї—М–љ—Л—Е —Б –Ї–ї–∞—Б—Б–Є—З–µ—Б–Ї–Є–Љ –і–µ–±—О—В–Њ–Љ (—Б –Љ—Л—И–µ—З–љ–Њ–є —Б–ї–∞–±–Њ—Б—В–Є), –Є –њ–Њ—Б–ї–µ–і—Г—О—Й–Є–Љ —А–∞–Ј–≤–Є—В–Є–µ–Љ –Ш–С–Ы, –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–µ –∞–љ—В–Є—В–µ–ї–∞ –њ—А–Є—Б—Г—В—Б—В–≤–Њ–≤–∞–ї–Є —В–Њ–ї—М–Ї–Њ –≤ 20% —Б–ї—Г—З–∞–µ–≤. –Я—А–µ–і—Б—В–∞–≤–ї—П–ї–∞ –Њ—Б–Њ–±—Л–є –Є–љ—В–µ—А–µ—Б –≥—А—Г–њ–њ–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –±–µ–Ј –љ–∞–ї–Є—З–Є—П –Љ–Є–Њ–Ј–Є—В–∞, –Њ–і–љ–∞–Ї–Њ, –Є–Љ–µ—О—Й–Є—Е —В—П–ґ–µ–ї–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е. –Т—Б–µ –±–Њ–ї—М–љ—Л–µ —Н—В–Њ–є –њ–Њ–і–≥—А—Г–њ–њ—Л –±—Л–ї–Є —Б–µ—А–Њ–њ–Њ–Ј–Є—В–Є–≤–љ—Л –њ–Њ –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–Љ –∞–љ—В–Є—В–µ–ї–∞–Љ, –љ–Њ –љ–Є–Ї—В–Њ –Є–Ј –љ–Є—Е –љ–µ –Є–Љ–µ–ї –∞–љ—В–Є—В–µ–ї–∞ –Ї Jo–1. –Р–≤—В–Њ—А—Л –Ј–∞–Ї–ї—О—З–∞—О—В, —З—В–Њ –љ–µ —В–Њ–ї—М–Ї–Њ Jo–1 –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–µ –∞–љ—В–Є—В–µ–ї–∞ –Љ–Њ–≥—Г—В –Є–і–µ–љ—В–Є—Д–Є—Ж–Є—А–Њ–≤–∞—В—М –њ–Њ–і–≥—А—Г–њ–њ—Л –≤–љ—Г—В—А–Є –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞.
–Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –Ь–°–Р, –≤ —З–∞—Б—В–љ–Њ—Б—В–Є, –∞–љ—В–Є—Б–Є–љ—В–µ—В–µ–Ј–љ—Л—Е –∞–љ—В–Є—В–µ–ї –њ—А–Њ–і–Њ–ї–ґ–∞—О—В—Б—П, —В–∞–Ї, –≤ 2005 –≥–Њ–і—Г Hashish L. —Б —Б–Њ–∞–≤—В. [14] —Б–Њ–Њ–±—Й–∞–µ—В –Њ–± –Њ–±–љ–∞—А—Г–ґ–µ–љ–Є–Є –љ–Њ–≤–Њ–≥–Њ (—Б–µ–і—М–Љ–Њ–≥–Њ) –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ–Њ–≥–Њ –∞–љ—В–Є—В–µ–ї–∞ – –Ї —В–Є—А–Њ–Ј–Є–ї – —В––†–Э–Ъ – —Б–Є–љ—В–µ—В–∞–Ј–µ —Г –±–Њ–ї—М–љ—Л—Е —Б –Я–Ь/–Ф–Ь. –Я–Њ–Ј–і–љ–µ–µ, –≤ 2007 –≥–Њ–і—Г Betteridge —Б —Б–Њ–∞–≤—В. [12] —Б—В–∞–ї–Њ –Є–Ј–≤–µ—Б—В–љ–Њ –Њ–± –Њ—В–Ї—А—Л—В–Є–Є, –µ—Й–µ –Њ–і–љ–Њ–≥–Њ (–≤–Њ—Б—М–Љ–Њ–≥–Њ) –∞–љ–∞—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ–Њ–≥–Њ –∞–љ—В–Є—В–µ–ї–∞ – –∞–љ—В–Є–Zo —Г –±–Њ–ї—М–љ—Л—Е —Б —Е–∞—А–∞–Ї—В–µ—А–љ—Л–Љ –і–ї—П –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞ —Б–Є–Љ–њ—В–Њ–Љ–Њ–Ї–Њ–Љ–њ–ї–µ–Ї—Б–Њ–Љ.
–Т 2008 –≥–Њ–і—Г Fathi M. —Б —Б–Њ–∞–≤—В. [15] –њ—А–Њ–≤–µ–і–µ–љ–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–Њ –Є–Ј—Г—З–µ–љ–Є—О –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Њ—З–љ–Њ–є —В–Ї–∞–љ–Є –њ—А–Є –Я–Ь/–Ф–Ь, –њ–Њ–і—В–≤–µ—А–і–Є–≤—И–µ–µ –≤—Л—Б–Њ–Ї—Г—О —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М –Ш–С–Ы (78%), –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ—Г—О —Б –ї–µ–≥–Њ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О –њ–Њ —А–µ—Б—В—А–Є–Ї—В–Є–≤–љ–Њ–Љ—Г —В–Є–њ—Г (—Б–љ–Є–ґ–µ–љ–Є–µ –Ц–Х–Ы).
–Ю–±—Б–ї–µ–і–Њ–≤–∞–љ–Њ 23 –њ–∞—Ж–Є–µ–љ—В–∞: 14 –±–Њ–ї—М–љ—Л—Е (9 –ґ–µ–љ—Й–Є–љ –Є 5 –Љ—Г–ґ—З–Є–љ) —Б –Я–Ь –Є 9 –ґ–µ–љ—Й–Є–љ —Б –Ф–Ь. –Т—Б–µ –±–Њ–ї—М–љ—Л–µ –њ–Њ–ї—Г—З–∞–ї–Є –≤—Л—Б–Њ–Ї–Є–µ –і–Њ–Ј—Л –У–Ъ —Б –Љ–µ–і–ї–µ–љ–љ—Л–Љ —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –≤ —В–µ—З–µ–љ–Є–µ –њ–µ—А–≤–Њ–≥–Њ –≥–Њ–і–∞ –±–Њ–ї–µ–Ј–љ–Є –Є –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є–≤–љ—Г—О —В–µ—А–∞–њ–Є—О (–∞–Ј–∞—В–Є–Њ–њ—А–Є–љ, –Љ–µ—В–Њ—В—А–µ–Ї—Б–∞—В, —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і, —Ж–Є–Ї–ї–Њ—Б–њ–Њ—А–Є–љ –Р –Є –≤–љ—Г—В—А–Є–≤–µ–љ–љ—Л–є –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ). –Ф–ї—П –Њ—Ж–µ–љ–Ї–Є –Ш–С–Ы –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–ї–Є—Б—М –ї–µ–≥–Њ—З–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–µ —В–µ—Б—В—Л (–±–Њ–і–Є–њ–ї—П—В–Є–Ј–Љ–Њ–≥—А–∞—Д–Є—П, –і–Є—Д—Д—Г–Ј–Є–Њ–љ–љ–∞—П —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –°–Ю (DL–°–Ю), –Ц–Х–Ы), —А–µ–љ—В–≥–µ–љ–Њ–≥—А–∞—Д–Є—П –Є –Ї–Њ–Љ–њ—М—О—В–µ—А–љ–∞—П —В–Њ–Љ–Њ–≥—А–∞—Д–Є—П –≤—Л—Б–Њ–Ї–Њ–≥–Њ —А–∞–Ј—А–µ—И–µ–љ–Є—П (–Ъ–Ґ–Т–†) –≥—А—Г–і–љ–Њ–є –Ї–ї–µ—В–Ї–Є.
–Э–∞ –Љ–Њ–Љ–µ–љ—В –њ–Њ—Б—В–∞–љ–Њ–≤–Ї–Є –і–Є–∞–≥–љ–Њ–Ј–∞, –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–∞—П –±–Њ–ї–µ–Ј–љ—М –ї–µ–≥–Ї–Є—Е (–Ш–С–Ы) –≤—Л—П–≤–ї–µ–љ–∞ —Г 18 –±–Њ–ї—М–љ—Л—Е (78%). –Э–Є —Г –Ї–Њ–≥–Њ –Є–Ј –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–µ –≤—Л—П–≤–ї–µ–љ–Њ –ї–µ–≥–Њ—З–љ–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є.
–§—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–µ –ї–µ–≥–Њ—З–љ—Л–µ —В–µ—Б—В—Л –њ—А–Њ–≤–µ–і–µ–љ—Л –≤ –љ–∞—З–∞–ї–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –Є –≤ –і–Є–љ–∞–Љ–Є–Ї–µ —Г 21 –±–Њ–ї—М–љ–Њ–≥–Њ, –Є 18 –±–Њ–ї—М–љ—Л—Е —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ. –Ґ–∞–Ї, —Г 8 –±–Њ–ї—М–љ—Л—Е (–Є–Ј 21) –≤—Л—П–≤–ї–µ–љ—Л —А–µ—Б—В—А–Є–Ї—В–Є–≤–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –ї–µ–≥–Њ—З–љ–Њ–є —В–Ї–∞–љ–Є (–Ц–Х–Ы –Љ–µ–љ–µ–µ 80%), –≤ —Б—А–µ–і–љ–µ–Љ, –Ц–Х–Ы —Б–Њ—Б—В–∞–≤–ї—П–ї–∞ 68%. –Т—Б–µ –њ–∞—Ж–Є–µ–љ—В—Л –Є–Љ–µ–ї–Є —Б–љ–Є–ґ–µ–љ–љ—Л–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є DL–°–Ю, –≤ —Б—А–µ–і–љ–µ–Љ, –љ–∞ 50%.
–С–Њ–ї—М–љ—Л–µ —Б –љ–∞–ї–Є—З–Є–µ–Љ –Ш–С–Ы –Є –±–µ–Ј —В–∞–Ї–Њ–≤–Њ–≥–Њ, –љ–µ —А–∞–Ј–ї–Є—З–∞–ї–Є—Б—М –њ–Њ –њ–Њ–ї—Г, –і–ї–Є—В–µ–ї—М–љ–Њ—Б—В–Є –±–Њ–ї–µ–Ј–љ–Є –Є –њ–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ –Љ–Є–Њ–Ј–Є—В–∞, –Њ–і–љ–∞–Ї–Њ —Г –±–Њ–ї—М–љ—Л—Е —Б –Ш–С–Ы –±—Л–ї–Є –±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Є–µ –Ј–љ–∞—З–µ–љ–Є—П –Ї—А–µ–∞—В–Є–љ—Д–Њ—Б—Д–Њ–Ї–Є–љ–∞–Ј—Л (–Ъ–§–Ъ) –і–Њ –ї–µ—З–µ–љ–Є—П. –Р–≤—В–Њ—А—Л –Њ—В–Љ–µ—З–∞—О—В, —З—В–Њ —Г –≤—Б–µ—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б Jo–1 –≤—Л—П–≤–ї–µ–љ—Л –њ—А–Є–Ј–љ–∞–Ї–Є –Ш–С–Ы.
–Ч–∞ –њ–µ—А–Є–Њ–і –љ–∞–±–ї—О–і–µ–љ–Є—П (35 –Љ–µ—Б—П—Ж–µ–≤), –і–≤–∞ –њ–∞—Ж–Є–µ–љ—В–∞ –њ–Њ–≥–Є–±–ї–Њ —З–µ—А–µ–Ј 10 –і–љ–µ–є –Є 18 –Љ–µ—Б—П—Ж–µ–≤ –Њ—В –Љ–Њ–Љ–µ–љ—В–∞ –њ–Њ—Б—В–∞–љ–Њ–≤–Ї–Є –і–Є–∞–≥–љ–Њ–Ј–∞. –Я—А–Є—З–Є–љ–Њ–є —Б–Љ–µ—А—В–Є —П–≤–Є–ї–Є—Б—М: –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–∞—П –ї–µ–≥–Њ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М —Б —А–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є —В—П–ґ–µ–ї–Њ–є –Ш–С–Ы –Є –≤—Л—Б–Њ–Ї–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –±–Њ–ї–µ–Ј–љ–Є, —А–µ–Ј–Є—Б—В–µ–љ—В–љ–∞—П –Ї –њ—А–Њ–≤–µ–і–µ–љ–Є—О —В–µ—А–∞–њ–Є–Є (–≤—Л—Б–Њ–Ї–Є–µ –і–Њ–Ј—Л –У–Ъ –≤ —Б–Њ—З–µ—В–∞–љ–Є–Є —Б –≤–љ—Г—В—А–Є–≤–µ–љ–љ—Л–Љ —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і–Њ–Љ). –£ –Њ–і–љ–Њ–є –њ–∞—Ж–Є–µ–љ—В–Ї–Є –љ–∞ –∞—Г—В–Њ–њ—Б–Є–Є –љ–∞–±–ї—О–і–∞–ї–Є—Б—М –њ–љ–µ–≤–Љ–Њ–љ–Є—П, —В–Њ—З–µ—З–љ—Л–µ –ї–µ–≥–Њ—З–љ—Л–µ –≥–µ–Љ–Њ—А—А–∞–≥–Є–Є –Є –Њ—Б—В—А—Л–є –Є–љ—Д–∞—А–Ї—В –ї–µ–≥–Ї–Њ–≥–Њ, —Г –і—А—Г–≥–Њ–є – –∞–ї—М–≤–µ–Њ–ї–Є—В. –£ –Њ—Б—В–∞–ї—М–љ—Л—Е (21 –њ–∞—Ж–Є–µ–љ—В) –љ–∞–±–ї—О–і–∞–ї–∞—Б—М –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–∞—П –і–Є–љ–∞–Љ–Є–Ї–∞ –њ—А–Њ—Ж–µ—Б—Б–∞ (—Б–љ–Є–ґ–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –±–Њ–ї–µ–Ј–љ–Є –Є —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –Њ–±—К–µ–Љ–∞ –і–≤–Є–ґ–µ–љ–Є–є, –љ–∞—А–∞—Б—В–∞–љ–Є–µ –Љ—Л—И–µ—З–љ–Њ–є —Б–Є–ї—Л –Є –љ–Њ—А–Љ–∞–ї–Є–Ј–∞—Ж–Є—П –Ъ–§–Ъ). –£ —В—А–µ—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Ј–∞ –њ–µ—А–Є–Њ–і –љ–∞–±–ї—О–і–µ–љ–Є—П –Њ—В–Љ–µ—З–µ–љ–Њ –Њ–±–Њ—Б—В—А–µ–љ–Є–µ –±–Њ–ї–µ–Ј–љ–Є. –≠—В–Є –њ–∞—Ж–Є–µ–љ—В—Л –Є–Љ–µ–ї–Є –±–Њ–ї–µ–µ –љ–Є–Ј–Ї–Є–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є –ї–µ–≥–Њ—З–љ—Л—Е —В–µ—Б—В–Њ–≤, –Є –±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ–љ—Л–µ —А–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, –±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Є–µ —Ж–Є—Д—А—Л –Ъ–§–Ъ, —З–µ–Љ –њ–∞—Ж–Є–µ–љ—В—Л –±–µ–Ј –Ш–С–Ы. –Ч–∞ –≤—А–µ–Љ—П –љ–∞–±–ї—О–і–µ–љ–Є—П –Ц–Х–Ы —Г–≤–µ–ї–Є—З–Є–ї–∞—Б—М —Г 33%, –Њ—Б—В–∞–ї–∞—Б—М —Б—В–∞–±–Є–ї—М–љ–Њ–є —Г 39% –Є —Г–Љ–µ–љ—М—И–Є–ї–∞—Б—М —Г 28%.
–Р–љ–∞–ї–Є–Ј–Є—А—Г—П —А–µ–Ј—Г–ї—М—В–∞—В—Л, –∞–≤—В–Њ—А—Л –њ—А–Є—Е–Њ–і—П—В –Ї —Б–ї–µ–і—Г—О—Й–Є–Љ –≤—Л–≤–Њ–і–∞–Љ:
1. –Ґ–µ—З–µ–љ–Є–µ –Ш–С–Ы —В—А—Г–і–љ–Њ –њ—А–Њ–≥–љ–Њ–Ј–Є—А–Њ–≤–∞—В—М –≤ –і–µ–±—О—В–µ –±–Њ–ї–µ–Ј–љ–Є, –њ–Њ—Н—В–Њ–Љ—Г –њ–∞—Ж–Є–µ–љ—В—Л —Б –Я–Ь/–Ф–Ь –Є –Ш–С–Ы –љ—Г–ґ–і–∞—О—В—Б—П –≤ —В—Й–∞—В–µ–ї—М–љ–Њ–Љ –љ–∞–±–ї—О–і–µ–љ–Є–Є, –≤–љ–Є–Љ–∞—В–µ–ї—М–љ–Њ–є –Њ—Ж–µ–љ–Ї–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Њ—Б—В–Њ—П–љ–Є—П, –ї–µ–≥–Њ—З–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е —В–µ—Б—В–Њ–≤ –Є —А–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ—Л.
2. –Ш–Ј–Љ–µ–љ–µ–љ–Є—П –Ц–Х–Ы –Є —А–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ—Л, –њ–Њ—П–≤–ї—П—О—Й–Є–µ—Б—П –њ–Њ—Б–ї–µ 11–12 –љ–µ–і–µ–ї–Є —В–µ—А–∞–њ–Є–Є, –Љ–Њ–≥—Г—В —Б–Њ—Е—А–∞–љ—П—В—М—Б—П –≤ —В–µ—З–µ–љ–Є–µ –і–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ –≤—А–µ–Љ–µ–љ–Є.
3. –Ш–С–Ы –њ—А–Є –Я–Ь/–Ф–Ь, –≤ –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤ –њ—А–Њ—В–µ–Ї–∞–µ—В —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є, –љ–Њ –Љ—П–≥–Ї–Њ –Є –љ–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г–µ—В –љ–∞ —Д–Њ–љ–µ –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є–≤–љ–Њ–є —В–µ—А–∞–њ–Є–Є.
4. –Я–Њ–Ї–∞–Ј–∞—В–µ–ї–Є –ї–µ–≥–Њ—З–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е —В–µ—Б—В–Њ–≤ –Љ–Њ–≥—Г—В –љ–Њ—А–Љ–∞–ї–Є–Ј–Њ–≤–∞—В—М—Б—П –Є–ї–Є —Г–ї—Г—З—И–∞—В—М—Б—П –љ–∞ —Д–Њ–љ–µ –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є–≤–љ–Њ–є —В–µ—А–∞–њ–Є–Є –і–∞–ґ–µ –≤ —В–Њ–Љ —Б–ї—Г—З–∞–µ, –µ—Б–ї–Є —А–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –љ–µ –Є–Ј–Љ–µ–љ—П–µ—В—Б—П.
5. –Т —В–Њ –ґ–µ –≤—А–µ–Љ—П, —В–µ—З–µ–љ–Є–µ, «–Њ—В–≤–µ—В» –љ–∞ —В–µ—А–∞–њ–Є—О –Є –њ—А–Њ–≥–љ–Њ–Ј –Ш–С–Ы –Љ–Њ–≥—Г—В –±—Л—В—М –≤–∞—А–Є–∞–±–µ–ї—М–љ—Л.
6. –Я–∞—Ж–Є–µ–љ—В—Л —Б –Я–Ь/–Ф–Ь –Є –Ш–С–Ы –Є–Љ–µ–µ—В —Е—Г–і—И–Є–є –њ—А–Њ–≥–љ–Њ–Ј. –†–∞–Ј–≤–Є—В–Є–µ –°–§–Р –Є–Љ–µ–µ—В –њ–ї–Њ—Е–Њ–є –њ—А–Њ–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–є –њ—А–Є–Ј–љ–∞–Ї –Є —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –≤—Л—Б–Њ–Ї–Њ–є –ї–µ—В–∞–ї—М–љ–Њ—Б—В—М—О.
7. –Я—А–Њ–≤–µ–і–µ–љ–Є–µ —А–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –ї–µ–≥–Ї–Є—Е –Є –Ъ–Ґ–Т–† –Њ—Б–Њ–±–µ–љ–љ–Њ –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Б –°–§–Р., –Њ–і–љ–∞–Ї–Њ, –Ъ–Ґ–Т–† —П–≤–ї—П–µ—В—Б—П –±–Њ–ї–µ–µ –Є–љ—Д–Њ—А–Љ–∞—В–Є–≤–љ—Л–Љ –Є —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ—Л–Љ –Љ–µ—В–Њ–і–Њ–Љ.
8. –Ы–µ–≥–Њ—З–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–µ —В–µ—Б—В—Л, —В–∞–Ї–ґ–µ, –і–Њ–±–∞–≤–ї—П—О—В –≤–∞–ґ–љ—Г—О –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—О, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Є—Е –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є —В–Њ–ї—М–Ї–Њ —З–∞—Б—В–Є—З–љ–Њ –Ї–Њ—А—А–µ–ї–Є—А—Г—О—В —Б –Ъ–Ґ–Т–† – —Б—З–µ—В–Њ–Љ –Є —А–Є—Б—Г–љ–Ї–Њ–Љ. –Я–Њ—Б–Ї–Њ–ї—М–Ї—Г DL–°–Ю –Њ–±—Л—З–љ–Њ —Б–љ–Є–ґ–µ–љ–∞ —Г –±–Њ–ї—М–љ—Л—Е —Б –Ш–С–Ы, –Є–ї–Є —Б–љ–Є–ґ–∞–µ—В—Б—П —Г –±–Њ–ї—М–љ—Л—Е —А–∞–љ—М—И–µ, —З–µ–Љ –Ц–Х–Ы, —В–Њ —Н—В–Њ—В –њ–Њ–Ї–∞–Ј–∞—В–µ–ї—М, —В–∞–Ї–ґ–µ, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –≤–Ї–ї—О—З–∞—В—М –≤ –њ–ї–∞–љ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –±–Њ–ї—М–љ—Л—Е –Я–Ь/–Ф–Ь.
9. –Ш–Ј–Њ–ї–Є—А–Њ–≤–∞–љ–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ DL–°–Ю – —А–∞–љ–љ–Є–є –њ—А–Є–Ј–љ–∞–Ї –Ш–С–Ы —Г –±–Њ–ї—М–љ—Л—Е —Б –Я–Ь/–Ф–Ь.
10. –Ш–Ј–Љ–µ–љ–µ–љ–Є—П –≤ –ї–µ–≥–Ї–Є—Е –њ—А–Є –Ъ–Ґ–Т–† –Љ–Њ–≥—Г—В —Б–Њ—Е—А–∞–љ—П—В—М—Б—П, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞ —А–µ–Љ–Є—Б—Б–Є—О –Љ—Л—И–µ—З–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞.
11. –£ –±–Њ–ї—М–љ—Л—Е —Б –Љ–∞–ї–Њ–≤—Л—А–∞–ґ–µ–љ–љ—Л–Љ —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –Љ—Л—И–µ—З–љ–Њ–є —Б–Є–ї—Л –Љ–Њ–ґ–µ—В —А–∞–Ј–≤–Є–≤–∞—В—М—Б—П –±—Л—Б—В—А–Њ–њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–∞—П –Ш–С–Ы.
12. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ј–љ–∞—З–Є–Љ–Њ—Б—В—М –±–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ–Њ–є –Ш–С–Ы —Г –±–Њ–ї—М–љ—Л—Е —Б –Я–Ь/–Ф–Ь –љ–µ –Њ—Ж–µ–љ–Є–≤–∞–µ—В—Б—П, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –љ–Є —Г –Њ–і–љ–Њ–≥–Њ –Є–Ј —В–∞–Ї–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –Ј–∞ –њ–µ—А–Є–Њ–і –љ–∞–±–ї—О–і–µ–љ–Є—П, –љ–µ –≤—Л—П–≤–ї–µ–љ—Л –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П.
–Я—А–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Д—Г–љ–Ї—Ж–Є–Є –≤–љ–µ—И–љ–µ–≥–Њ –і—Л—Е–∞–љ–Є—П –∞–≤—В–Њ—А—Л –Њ–±—А–∞—Й–∞—О—В –≤–љ–Є–Љ–∞–љ–Є–µ –љ–∞ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –ї–µ–≥–Њ—З–љ—Л—Е –Њ–±—К–µ–Љ–Њ–≤ –Є —А–µ–Ј–µ—А–≤–Њ–≤ –і—Л—Е–∞–љ–Є—П.
–Р–≤—В–Њ—А—Л, —В–∞–Ї–ґ–µ, –≤—Л–і–µ–ї—П—О—В —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –і–ї—П –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Ї–Є—Е –њ—А–Є –Я–Ь/–Ф–Ь –Ъ–Ґ––Є–Ј–Љ–µ–љ–µ–љ–Є—П: –≤ –≤–Є–і–µ —В—П–ґ–Є—Б—В–Њ–є –Є —П—З–µ–Є—Б—В–Њ–є –і–µ—Д–Њ—А–Љ–∞—Ж–Є–Є –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —А–Є—Б—Г–љ–Ї–∞ –њ–Њ —В–Є–њ—Г “—Б–Њ—В–Њ–≤–Њ–≥–Њ –ї–µ–≥–Ї–Њ–≥–Њ”
–Я—А–µ–і—Б—В–∞–≤–ї—П—О—В –Њ—Б–Њ–±—Л–є –Є–љ—В–µ—А–µ—Б —А–µ–Ј—Г–ї—М—В–∞—В—Л, –њ–Њ–ї—Г—З–µ–љ–љ—Л–µ –∞–≤—В–Њ—А–∞–Љ–Є –њ—А–Є –Є–Ј—Г—З–µ–љ–Є–Є —А–Њ–ї–Є –∞–љ—В–Є–Jo–1 –∞–љ—В–Є—В–µ–ї. –°–Њ–≥–ї–∞—Б–љ–Њ –Њ–±—Й–µ–њ—А–Є–љ—П—В–Њ–Љ—Г —А–∞–љ–µ–µ –Љ–љ–µ–љ–Є—О, –Є—Е –≤—Л—П–≤–ї–µ–љ–Є–µ —П–≤–ї—П–µ—В—Б—П –њ–ї–Њ—Е–Є–Љ –њ—А–Њ–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ. –Ю–і–љ–∞–Ї–Њ, –≤ –і–∞–љ–љ–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є, –∞–љ—В–Є– Jo–1 –≤—Л—П–≤–ї—П–ї–Є—Б—М —Г –±–Њ–ї—М–љ—Л—Е, –Ї–Њ—В–Њ—А—Л–µ –±—Л–ї–Є —Б—В–∞–±–Є–ї—М–љ—Л –Є–ї–Є —Г –Ї–Њ—В–Њ—А—Л—Е –Њ—В–Љ–µ—З–∞–ї–Њ—Б—М —Г–ї—Г—З—И–µ–љ–Є–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –ї–µ–≥–Њ—З–љ—Л—Е —В–µ—Б—В–Њ–≤. –Т —В–Њ –ґ–µ –≤—А–µ–Љ—П —Г –±–Њ–ї—М–љ—Л—Е —Б –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ–Њ–є –і–Є–љ–∞–Љ–Є–Ї–Њ–є –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –Њ–љ–Є –љ–µ –≤—Л—П–≤–ї—П–ї–Є—Б—М. –Р–≤—В–Њ—А—Л —Б—З–Є—В–∞—О—В, —З—В–Њ –∞–љ—В–Є–Jo–1 –∞–љ—В–Є—В–µ–ї–∞ —П–≤–ї—П—О—В—Б—П –љ–µ —В–Њ–ї—М–Ї–Њ –њ—А–µ–і–Є–Ї—В–Њ—А–∞–Љ–Є –Ш–С–Ы, –љ–Њ –Є –Њ–±–ї–∞–і–∞—О—В –њ–Њ–Ј–Є—В–Є–≤–љ—Л–Љ –њ—А–Њ–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–Љ –Ј–љ–∞—З–µ–љ–Є–µ–Љ.
–Я–Њ–і–Њ–±–љ–Њ–µ –Љ–љ–µ–љ–Є–µ –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ —А–Њ–ї–Є –∞–љ—В–Є–Jo–1 –∞–љ—В–Є—В–µ–ї –љ–µ —В–Њ–ї—М–Ї–Њ –љ–µ –њ—А–Њ—В–Є–≤–Њ—А–µ—З–Є—В, –∞, –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, –Є –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В –њ—А–Є–≤–µ–і–µ–љ–љ—Л–µ –≤—Л—И–µ (Yoshifuji —Б —Б–Њ–∞–≤—В.) –Њ–њ–Є—Б–∞–љ–Є—П —Б–ї—Г—З–∞–µ–≤ —В—П–ґ–µ–ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Ї–Є—Е —Г –±–Њ–ї—М–љ—Л—Е —Б –Љ–Є–љ–Є–Љ–∞–ї—М–љ—Л–Љ–Є –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є –Љ–Є–Њ–Ј–Є—В–∞, –њ–Њ–Ј–Є—В–Є–≤–љ—Л—Е –њ–Њ –∞–љ—В–Є—Б–Є–љ—В–µ—В–∞–Ј–љ—Л–Љ –∞–љ—В–Є—В–µ–ї–∞–Љ, –≤ –Њ—В—Б—Г—В—Б—В–≤–Є–Є J–Њ–1.
–Ш–љ—В–µ—А–µ—Б–љ–Њ —В–∞–Ї–ґ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ, –њ—А–Њ–≤–µ–і–µ–љ–љ–Њ–µ –≤ 2005 –≥–Њ–і—Г —П–њ–Њ–љ—Б–Ї–Є–Љ–Є –Ї–Њ–ї–ї–µ–≥–∞–Љ–Є Fujisawa T —Б —Б–Њ–∞–≤—В. [16] –њ–Њ –Є–Ј—Г—З–µ–љ–Є—О —А–∞–Ј–ї–Є—З–Є—П —В–µ—З–µ–љ–Є—П –Є –њ—А–Њ–≥–љ–Њ–Ј–∞ –ї–µ–≥–Њ—З–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ—А–Є –Я–Ь –Є –Ф–Ь. –Ю–±—Б–ї–µ–і–Њ–≤–∞–љ–Њ 28 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ (16 – —Б –Я–Ь –Є 12 – —Б –Ф–Ь), —Г –≤—Б–µ—Е –њ—А–Є—Б—Г—В—Б—В–≤–Њ–≤–∞–ї–Є –њ—А–Є–Ј–љ–∞–Ї–Є –Ш–С–Ы. –Я—А–Њ–≤–µ–і–µ–љ—Л: –Ъ–Ґ––Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ, –±—А–Њ–љ—Е–Њ–∞–ї—М–≤–µ–Њ–ї—П—А–љ—Л–є –ї–∞–≤–∞–ґ (–С–Р–Ы), –±–Є–Њ–њ—Б–Є—П –ї–µ–≥–Ї–Є—Е (—Г 5 –±–Њ–ї—М–љ—Л—Е —Б –Я–Ь, 5 – —Б –Ф–Ь –Є —Г 3–—Е –њ–Њ–≥–Є–±—И–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ – –∞—Г—В–Њ–њ—Б–Є—П). –Ю–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–Њ–Ї–∞–Ј–∞–ї–Њ, —З—В–Њ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤, —Б—В—А–∞–і–∞—О—Й–Є—Е –Ф–Ь –±—Л–ї–Є —Е—Г–і—И–Є–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л –Є –≤—Л–ґ–Є–≤–∞–µ–Љ–Њ—Б—В—М. –Ґ–∞–Ї, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞ –±–Њ–ї–µ–µ –љ–Є–Ј–Ї–Є–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є –Ъ–§–Ъ —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Ф–Ь, –Њ—В–Љ–µ—З–∞–ї–∞—Б—М –±–Њ–ї—М—И–∞—П —А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В—М –Ї –У–Ъ, —З–∞—Й–µ –≤—Б—В—А–µ—З–∞–ї—Б—П –і–Є—Д—Д—Г–Ј–љ—Л–є –∞–ї—М–≤–µ–Њ–ї–Є—В (—Г 3 –њ–Њ–≥–Є–±—И–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ – 37%), –≤—Л—П–≤–ї—П–ї–Њ—Б—М –±–Њ–ї—М—И–µ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –љ–µ–є—В—А–Њ—Д–Є–ї–Њ–≤ (–њ–ї–Њ—Е–Њ–є –њ—А–Њ–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–є –њ—А–Є–Ј–љ–∞–Ї), –ї–Є–Љ—Д–Њ—Ж–Є—В–Њ–≤ –Є —Н–Њ–Ј–Є–љ–Њ—Д–Є–ї–Њ–≤ –њ—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–µ –С–Р–Ы. –Ґ–Њ–≥–і–∞ –Ї–∞–Ї —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤, —Б—В—А–∞–і–∞—О—Й–Є—Е –Я–Ь —З–∞—Й–µ –љ–∞–±–ї—О–і–∞–ї–∞—Б—М –љ–µ—Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–∞—П –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–∞—П –њ–љ–µ–≤–Љ–Њ–љ–Є—П (80%) –Є –љ–Є —Г –Њ–і–љ–Њ–≥–Њ –±–Њ–ї—М–љ–Њ–≥–Њ —Б –Я–Ь –љ–µ –≤—Л—П–≤–ї–µ–љ–Њ –і–Є—Д—Д—Г–Ј–љ–Њ–≥–Њ –∞–ї—М–≤–µ–Њ–ї–Є—В–∞. –Ґ–∞–Ї, —Д–∞–Ї—В–Њ—А–∞–Љ–Є –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ–Њ–≥–Њ –њ—А–Њ–≥–љ–Њ–Ј–∞, –Ї–∞–Ї –Є –≤ –њ—А–µ–і—Л–і—Г—Й–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е, –∞–≤—В–Њ—А—Л –≤—Л–і–µ–ї—П—О—В –і–Є—Д—Д—Г–Ј–љ—Л–є –∞–ї—М–≤–µ–Њ–ї–Є—В, –±–Њ–ї—М—И–Њ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –љ–µ–є—В—А–Њ—Д–Є–ї–Њ–≤ –њ—А–Є –С–Р–Ы, –∞ —В–∞–Ї–ґ–µ –љ–Є–Ј–Ї–Є–µ —Ж–Є—Д—А—Л –Ъ–§–Ъ —Г –±–Њ–ї—М–љ—Л—Е —Б –Ш–С–Ы. –Т –ї–µ—З–µ–љ–Є–Є –±–Њ–ї—М–љ—Л—Е –Я–Ь/–Ф–Ь —Б –Ш–С–Ы –∞–≤—В–Њ—А—Л –љ–∞–±–ї—О–і–∞—О—В –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–є —Н—Д—Д–µ–Ї—В –њ—А–Є –њ—А–Є–Љ–µ–љ–µ–љ–Є–Є —Ж–Є–Ї–ї–Њ—Б–њ–Њ—А–Є–љ–∞ –Р, —Ж–Є–Ї–ї–Њ—Д–Њ—Б—Д–∞–Љ–Є–і–∞ –Є –≤–љ—Г—В—А–Є–≤–µ–љ–љ–Њ–≥–Њ –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ –≤ —Б–Њ—З–µ—В–∞–љ–Є–Є —Б –≤—Л—Б–Њ–Ї–Є–Љ–Є –і–Њ–Ј–∞–Љ–Є –У–Ъ.
–Ю–±—Б—Г–ґ–і–∞—П –њ–Њ–ї—Г—З–µ–љ–љ—Л–µ –і–∞–љ–љ—Л–µ, –∞–≤—В–Њ—А—Л –њ—А–µ–і–њ–Њ–ї–∞–≥–∞—О—В, —З—В–Њ –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П –њ—А–Є –Ф–Ь –Є –Я–Ь —А–∞–Ј–ї–Є—З–љ—Л, —В–Њ, –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ –Є —А–∞–Ј–ї–Є—З–µ–љ –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Њ—З–љ–Њ–є —В–Ї–∞–љ–Є.
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –≤ —Б–≤—П–Ј–Є —Б –≤—Л—Б–Њ–Ї–Њ–є —З–∞—Б—В–Њ—В–Њ–є –Ш–С–Ы –њ—А–Є –Я–Ь/–Ф–Ь, —А–µ–Ї–Њ–Љ–µ–љ–і–Њ–≤–∞–љ–Њ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –±–Њ–ї—М–љ—Л—Е, –≤–Ї–ї—О—З–∞—О—Й–µ–µ –ї–µ–≥–Њ—З–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ—Л–µ —В–µ—Б—В—Л, —А–µ–љ—В–≥–µ–љ–Њ–≥—А–∞–Љ–Љ—Л –≥—А—Г–і–љ–Њ–є –Ї–ї–µ—В–Ї–Є –Є –Ъ–Ґ –і–ї—П –≤—Л—П–≤–ї–µ–љ–Є—П –Ш–С–Ы –љ–∞ —А–∞–љ–љ–Є—Е —Б—В–∞–і–Є—П—Е –±–Њ–ї–µ–Ј–љ–Є, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Є –њ–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е –Є –Љ–Є–Њ–њ–∞—В–Є—П –Љ–Њ–≥—Г—В –≤–ї–Є—П—В—М –љ–∞ –µ–ґ–µ–і–љ–µ–≤–љ—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –±–Њ–ї—М–љ–Њ–≥–Њ –Є –Ї–∞—З–µ—Б—В–≤–Њ –µ–≥–Њ –ґ–Є–Ј–љ–Є.
–Я—А–Є –њ–Њ—Б—В–∞–љ–Њ–≤–Ї–µ –і–Є–∞–≥–љ–Њ–Ј–∞ –Ш–С–Ы –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Њ–µ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–∞—Ж–Є–µ–љ—В–∞ –і–ї—П –Њ—Ж–µ–љ–Ї–Є –њ—А–Њ–≥–љ–Њ–Ј–∞ –±–Њ–ї–µ–Ј–љ–Є. –•–Њ—В—П —В–µ—З–µ–љ–Є–µ –±–Њ–ї–µ–Ј–љ–Є, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞ –њ—А–Њ–≤–µ–і–µ–љ–Є–µ –Є–Љ–Љ—Г–љ–Њ—Б—Г–њ—А–µ—Б—Б–Є–≤–љ–Њ–є —В–µ—А–∞–њ–Є–Є, –Љ–Њ–ґ–µ—В –≤–∞—А—М–Є—А–Њ–≤–∞—В—М, –≤ –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ —Б–ї—Г—З–∞–µ–≤, –њ—А–Њ–≥–љ–Њ–Ј –±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–є —Б–Њ —Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є–µ–є, –Є–ї–Є –і–∞–ґ–µ –љ–Њ—А–Љ–∞–ї–Є–Ј–∞—Ж–Є–µ–є –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –ї–µ–≥–Њ—З–љ—Л—Е –Њ–±—К–µ–Љ–Њ–≤ –Є –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–∞—П –і–Є–љ–∞–Љ–Є–Ї–∞ –ї–µ–≥–Њ—З–љ—Л—Е –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –Ї–Њ—А—А–µ–ї–Є—А—Г–µ—В —Б –љ–∞—А–∞—Б—В–∞–љ–Є–µ–Љ –Љ—Л—И–µ—З–љ–Њ–є —Б–Є–ї—Л.
–Э–µ–Њ–±—Е–Њ–і–Є–Љ—Л –і–∞–ї—М–љ–µ–є—И–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –і–ї—П –Њ–њ—А–µ–і–µ–ї–µ–љ–љ–Њ–≥–Њ –Ј–∞–Ї–ї—О—З–µ–љ–Є—П –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ –њ—А–Њ–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Њ–є —А–Њ–ї–Є –∞–љ—В–Є–Jo–1 –∞–љ—В–Є—В–µ–ї.


–Ы–Є—В–µ—А–∞—В—Г—А–∞
1. Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet 2003, 362, 971.
2. Plotz, PH, Dalakas, M, Leff, RL, et al. Current concepts in the idiopathic inflammatory myopathies: Polymyositis, dermatomyositis and related disorders. Ann Intern Med, 1989; 111–143.
3. Bohan, A, Peter, JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975; 292:403.
4. Bohan, A, Peter, JB, Bowman, RL, Pearson, CM. Computer–assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255.
5. –Ш–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–µ –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–µ –Љ–Є–Њ–њ–∞—В–Є–Є. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ —А–µ–Ї–Њ–Љ–µ–љ–і–∞—Ж–Є–Є. –†–µ–≤–Љ–∞—В–Њ–ї–Њ–≥–Є—П. –У–ї.—А–µ–і. —З–ї.––Ї–Њ—А—А. –†–Р–Ь–Э –Х.–Ы.–Э–∞—Б–Њ–љ–Њ–≤. –Ь–Њ—Б–Ї–≤–∞ 2005, 192–201.
6. –Э–∞—Б–Њ–љ–Њ–≤ –Х.–Ы., –®—В—Г—В–Љ–∞–љ –Т.–Ч., –°–∞–ї–Њ–ґ–Є–љ –Ъ.–Т, –У—Г—Б–µ–≤–∞ –Э.–У., –Э–∞—Б–Њ–љ–Њ–≤–∞ –Т.–Р, –Я–ї–Њ—В—Ж –Я. –Ъ–ї–Є–љ–Є–Ї–Њ – –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –≥–µ—В–µ—А–Њ–≥–µ–љ–љ–Њ—Б—В—М –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є—Е –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Љ–Є–Њ–њ–∞—В–Є–є. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ь–µ–і–Є—Ж–Є–љ–∞ 1995, 2, 3–8.
7. –Р–љ—В–µ–ї–∞–≤–∞ –Ю.–Р., –°–Њ–ї–Њ–≤—М–µ–≤ –°.–Ъ., –•–Є—В—А–Њ–≤ –Р.–Э., –Э–∞—Б–Њ–љ–Њ–≤ –Х.–Ы. –Э–Њ–≤—Л–µ –∞—Б–њ–µ–Ї—В—Л —Д–∞—А–Љ–∞–Ї–Њ—В–µ—А–∞–њ–Є–Є –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є—Е –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –Љ–Є–Њ–њ–∞—В–Є–є (–Ю–±–Ј–Њ—А –ї–Є—В–µ—А–∞—В—Г—А—Л). –†—Г—Б—Б–Ї–Є–є –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Є–є –ґ—Г—А–љ–∞–ї 2006, 14, 8, 260, 627–629.
8. M–∞rir I.E. Hachulla, P. CH.Erin, S. Interstitial Lung Disease in Polymyositis and Dermatomyositis Arthritis & Rheumatism (Arthritis Care & Research) Vol. 47, 6, 15, 2002, 614–622.
9. –С–Њ–љ–і–∞—А–µ–љ–Ї–Њ –Ш.–С., –Ь—Г—Е–Є–љ –Э.–Р., –Э–∞—Б–Њ–љ–Њ–≤ –Х.–Ы. –Я–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е –њ—А–Є –њ–Њ–ї–Є–Љ–Є–Њ–Ј–Є—В–µ –Є –і–µ—А–Љ–∞—В–Њ–Љ–Є–Њ–Ј–Є—В–µ. –Ш–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –ї–µ–≥–Ї–Є—Е. –†—Г–Ї–Њ–≤–Њ–і—Б—В–≤–Њ –і–ї—П –≤—А–∞—З–µ–є. –Я–Њ–і —А–µ–і–∞–Ї—Ж–Є–µ–є –Ш–ї—М–Ї–Њ–≤–Є—З–∞ –Ь.–Ь. –Ъ–Њ–Ї–Њ—Б–Њ–≤–∞ –Р.–Э. 274–287. –°–∞–љ–Ї—В––Я–µ—В–µ—А—Г—А–≥. –Э–Њ—А–і–Љ–µ–і–Є–Ј–і–∞—В 2005.
10. OLeary P.A., Waisman M. Dermatomyositis: A stady of forty cases// Arch.Dermatol.Syph.– 1940.– вДЦ 41. – –†. 1001–1019.
11. –Ю–≤—З–∞—А–µ–љ–Ї–Њ –°.–Ш., –Я–µ—В—Г—Е–Њ–≤–∞ –Э.–Т., –°–ї—Г–Ї–Є–љ–∞ –Ю.–У. –Я–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е –њ–Њ —В–Є–њ—Г –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–є –±–Њ–ї–µ–Ј–љ–Є —Г –±–Њ–ї—М–љ–Њ–є —Б –і–µ—А–Љ–∞—В–Њ–Љ–Є–Њ–Ј–Є—В–Њ–Љ// –Э–Њ–≤—Л–µ –°–∞–љ–Ї—В––Я–µ—В–µ—А–±—Г—А–≥—Б–Ї–Є–µ –≤—А–∞—З–µ–±–љ—Л–µ –≤–µ–і–Њ–Љ–Њ—Б—В–Є. – 2001. –вДЦ 2– –°.49–51.
12. Betteridge Z., Gunawardena H., North J., Slinn J., McHugh N. Anti–synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti–Zo) assotiated with polymyositis and interstitial pneumonia. Rheumatol. 2007, 46, 1005–1008.
13. Yoshifuji H, Fujii T, Kobayashi S et al. Anti–aminoacyl–tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006, 39, 233–41.
14. Hashish L , Trieu EP, Sadanandan P, Targoff IN. Identification of autoantibodies to tyrosyl–tRNA synthetase in dermatomyositis with features consistent with antisynthetase syndrome (absract). Arthritis Rheum. 2005, 52, Suppl.9, 312.
15. Fathi –Ь, Vikgren J, Boijsen M, Tylen U, Jorfeldt L. et al Interstitial lung disease in Polymyositis and Dermatomyositis: longitudinal evaluation by pulmonary function and radiology. Arthritis and Rheumatusm (Arthritis Care and Research) Vol.59, No.5, May 15, 2008, 677–685.
16. Fujisawa T., I SUDA –Ґ, NAKAMURA Y., ENOMOTO N, IDE K., TOYOSHIMA M., UCHIYAMA H., et al. Differences in Clinical Features and Prognosis of Interstitial Lung Diseases Between Polymyositis and Dermatomyositis. The Journal of Rheumatology 2005; 32,1, 58–64.



