



.gif)
–Ч–∞ –њ–Њ—Б–ї–µ–і–љ–Є–µ –і–µ—Б—П—В–Є–ї–µ—В–Є—П –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Є—П –Њ–± –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ —А–∞–і–Є–Ї–∞–ї—М–љ–Њ –Є–Ј–Љ–µ–љ–Є–ї–Є—Б—М, —З—В–Њ —Б–≤—П–Ј–∞–љ–Њ —Б —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є–µ–Љ –≥–µ—В–µ—А–Њ–≥–µ–љ–љ–Њ—Б—В–Є –±–µ–ї–Ї–Њ–≤–Њ–≥–Њ —Б–Њ—Б—В–∞–≤–∞ –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е —Д–Є–±—А–Є–ї–ї. –Э–∞ –Њ—Б–љ–Њ–≤–∞–љ–Є–Є —Б–њ–µ—Ж–Є—Д–Є—З–љ–Њ—Б—В–Є –±–µ–ї–Ї–∞ –∞–Љ–Є–ї–Њ–Є–і–∞ —Б—В—А–Њ–Є—В—Б—П —Б–Њ–≤—А–µ–Љ–µ–љ–љ–∞—П –Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є—П. –†–∞–Ј–і–µ–ї—П—О—В —Б–Є—Б—В–µ–Љ–љ—Л–µ –Є –ї–Њ–Ї–∞–ї—М–љ—Л–µ —Д–Њ—А–Љ—Л –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞.
–†–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М –РL––∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –њ–Њ –і–∞–љ–љ—Л–Љ –Э–∞—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–≥–Њ —Ж–µ–љ—В—А–∞ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–є —Б—В–∞—В–Є—Б—В–Є–Ї–Є –°–®–Р, —Б–Њ—Б—В–∞–≤–ї—П–µ—В 4,5 —Б–ї—Г—З–∞—П –љ–∞ 100000 [1]. –†–Њ—Б—В –Ј–∞–±–Њ–ї–µ–≤–∞–µ–Љ–Њ—Б—В–Є, —Б—В–∞–љ–і–∞—А—В–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л–є –њ–Њ –≤–Њ–Ј—А–∞—Б—В—Г, —Б–Њ—Б—В–∞–≤–ї—П–µ—В 5,1–12,8 –љ–∞ –Љ–Є–ї–ї–Є–Њ–љ —З–µ–ї–Њ–≤–µ–Ї–Њ––ї–µ—В, —З—В–Њ —Б–Њ—Б—В–∞–≤–ї—П–µ—В –њ—А–Є–Љ–µ—А–љ–Њ 3200 –љ–Њ–≤—Л—Е —Б–ї—Г—З–∞–µ–≤ –≤ –≥–Њ–і –≤ –°–®–Р [2].
–Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —Б—З–Є—В–∞–µ—В—Б—П, —З—В–Њ –≤ –Њ—Б–љ–Њ–≤–µ —А–∞–Ј–≤–Є—В–Є—П –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –ї–µ–ґ–Є—В –і–Њ–±—А–Њ–Ї–∞—З–µ—Б—В–≤–µ–љ–љ–∞—П –њ–ї–∞–Ј–Љ–∞–Ї–ї–µ—В–Њ—З–љ–∞—П –і–Є—Б–Ї—А–∞–Ј–Є—П –Ї–Њ—Б—В–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –Р–љ–Њ–Љ–∞–ї—М–љ—Л–є –Ї–ї–Њ–љ –њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї –њ—А–Њ–і—Г—Ж–Є—А—Г–µ—В –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ—Л–µ –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ—Л, –њ—А–µ–і—Б—В–∞–≤–ї—П—О—Й–Є–µ –ї–µ–≥–Ї–Є–µ —Ж–µ–њ–Є –Љ–Њ–љ–Њ–Ї–ї–Њ–љ–∞–ї—М–љ–Њ–≥–Њ –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞, —З–∞—Й–µ –ї—П–Љ–±–і–∞ (–ї), —А–µ–ґ–µ –Ї–∞–њ–њ–∞–—В–Є–њ–∞ (k) (–Є—Е —Б–Њ–Њ—В–љ–Њ—И–µ–љ–Є–µ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 3:1). –Р–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В–љ—Л–є –∞–љ–∞–ї–Є–Ј –њ–Њ–Ї–∞–Ј—Л–≤–∞–µ—В, —З—В–Њ –љ–µ–Ї–Њ—В–Њ—А—Л–µ –∞–Љ–Є–љ–Њ–Ї–Є—Б–ї–Њ—В—Л –≤ –≤–∞—А–Є–∞–±–µ–ї—М–љ—Л—Е —Г—З–∞—Б—В–Ї–∞—Е –ї–µ–≥–Ї–Є—Е —Ж–µ–њ–µ–є —Н—В–Є—Е –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–Њ–≤ –Ј–∞–љ–Є–Љ–∞—О—В –љ–µ–Њ–±—Л—З–љ—Г—О –њ–Њ–Ј–Є—Ж–Є—О, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї –љ–µ—Б—В–∞–±–Є–ї—М–љ–Њ—Б—В–Є –Є —Б–Ї–ї–Њ–љ–љ–Њ—Б—В–Є –Ї —Д–Є–±—А–Є–ї–ї–Њ–≥–µ–љ–µ–Ј—Г. –Ю—В–Љ–µ—З–µ–љ–Њ, —З—В–Њ –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –≤—Б—В—А–µ—З–∞–µ–Љ—Л–µ –њ–Њ–і–≥—А—Г–њ–њ—Л –ї–µ–≥–Ї–Є—Е —Ж–µ–њ–µ–є –≤ –РL––і–µ–њ–Њ–Ј–Є—В–∞—Е –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ—Л –ї VI –Є I —В–Є–њ–∞–Љ–Є. –≠—В–Њ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ—А–µ–і–њ–Њ–ї–∞–≥–∞—В—М, —З—В–Њ –Є–Љ–µ–љ–љ–Њ –Њ–љ–Є —Б—З–Є—В–∞—О—В—Б—П –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–љ—Л–Љ–Є. –Т –і–∞–ї—М–љ–µ–є—И–µ–Љ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ—А–Њ—Ж–µ—Б—Б –њ—А–µ—Ж–Є–њ–Є—В–∞—Ж–Є–Є –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е –±–µ–ї–Ї–Њ–≤ –≤ —В–Ї–∞–љ—П—Е –Њ—А–≥–∞–љ–Њ–≤––Љ–Є—И–µ–љ–µ–є [3]. –Ю—Б–љ–Њ–≤–љ—Л–µ –Њ—А–≥–∞–љ—Л––Љ–Є—И–µ–љ–Є –њ—А–Є –РL––∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ – —Б–µ—А–і—Ж–µ, –њ–Њ—З–Ї–Є, –ґ–µ–ї—Г–і–Њ—З–љ–Њ––Ї–Є—И–µ—З–љ—Л–є —В—А–∞–Ї—В, —А–µ—Б–њ–Є—А–∞—В–Њ—А–љ–∞—П —Б–Є—Б—В–µ–Љ–∞ –Є –і—А—Г–≥–Є–µ –Њ—А–≥–∞–љ—Л.
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Љ–љ–Њ–≥–Њ–Њ–±—А–∞–Ј–љ–∞ –Є –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–∞ –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —В–µ—Е –Є–ї–Є –Є–љ—Л—Е –Њ—А–≥–∞–љ–Њ–≤. –Я–µ—А–≤—Л–Љ–Є —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —П–≤–ї—П—О—В—Б—П —Б–ї–∞–±–Њ—Б—В—М –Є —Б–љ–Є–ґ–µ–љ–Є–µ –≤–µ—Б–∞. –Я–Њ—А–∞–ґ–µ–љ–Є–µ —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–Є—Б—В–µ–Љ—Л, –њ–Њ –і–∞–љ–љ—Л–Љ –Ю.–Ь. –Т–Є–љ–Њ–≥—А–∞–і–Њ–≤–Њ–є (1980), –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є –≤—Б—В—А–µ—З–∞–µ—В—Б—П —Г 70% –±–Њ–ї—М–љ—Л—Е, –њ—А–Є –∞–љ–∞—В–Њ–Љ–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є – –≤ 87% —Б–ї—Г—З–∞–µ–≤. –Я–∞—В–Њ–ї–Њ–≥–Є—П —Б–µ—А–і—Ж–∞ —П–≤–ї—П–µ—В—Б—П –≤–µ–і—Г—Й–Є–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є–µ–Љ –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Є –≤ –њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –Љ–Є–Њ–Ї–∞—А–і–∞. –Р–Љ–Є–ї–Њ–Є–і –Њ—В–Ї–ї–∞–і—Л–≤–∞–µ—В—Б—П –Љ–µ–ґ–Љ—Л—И–µ—З–љ–Њ, –њ–µ—А–Є–≤–∞—Б–Ї—Г–ї—П—А–љ–Њ, –≤—Л–Ј—Л–≤–∞—П –∞—В—А–Њ—Д–Є—О –Љ—Л—И–µ—З–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ. –Ь–Є–Њ–Ї–∞—А–і —В–µ—А—П–µ—В —Б–≤–Њ—О —Н–ї–∞—Б—В–Є—З–љ–Њ—Б—В—М, —Б—В–∞–љ–Њ–≤–Є—В—Б—П —А–Є–≥–Є–і–љ—Л–Љ, –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —З–µ–≥–Њ —Б—В—А–∞–і–∞–µ—В —Д—Г–љ–Ї—Ж–Є—П –і–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–≥–Њ —А–∞—Б—Б–ї–∞–±–ї–µ–љ–Є—П. –Я–Њ–ї–Њ—Б—В–Є –ї–µ–≤–Њ–≥–Њ –Є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –љ–µ –і–Є–ї–∞—В–Є—А—Г—О—В—Б—П, –∞ –њ–Њ–ї–Њ—Б—В–Є –њ—А–µ–і—Б–µ—А–і–Є–є —А–µ–Ј–Ї–Њ —А–∞—Б—И–Є—А—П—О—В—Б—П. –†–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –Ї–∞—А–і–Є–Њ–Љ–µ–≥–∞–ї–Є—П, –≤–њ–ї–Њ—В—М –і–Њ cor bovinum. –£–≤–µ–ї–Є—З–µ–љ–Є–µ –Љ–∞—Б—Б—Л –Љ–Є–Њ–Ї–∞—А–і–∞ –±—Л–≤–∞–µ—В –љ–∞—Б—В–Њ–ї—М–Ї–Њ –≤—Л—А–∞–ґ–µ–љ–љ—Л–Љ, —З—В–Њ –љ–∞ –≤—Б–Ї—А—Л—В–Є–Є —Б–µ—А–і—Ж–µ –Ј–∞–љ–Є–Љ–∞–µ—В –±–Њ–ї—М—И—Г—О —З–∞—Б—В—М –≥—А—Г–і–љ–Њ–є –Ї–ї–µ—В–Ї–Є (–Љ–∞—Б—Б–∞ —Б–µ—А–і—Ж–∞ –Љ–Њ–ґ–µ—В –і–Њ—Е–Њ–і–Є—В—М –і–Њ 1 –Ї–≥). –Ф–µ–њ–Њ–Ј–Є—В—Л –∞–Љ–Є–ї–Њ–Є–і–∞ –Љ–Њ–≥—Г—В —А–∞—Б–њ–Њ–ї–∞–≥–∞—В—М—Б—П –≤ –њ—А–Њ–≤–Њ–і—П—Й–µ–є —Б–Є—Б—В–µ–Љ–µ —Б–µ—А–і—Ж–∞, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї —А–∞–Ј–ї–Є—З–љ—Л–Љ –љ–∞—А—Г—И–µ–љ–Є—П–Љ —А–Є—В–Љ–∞ –Є –Љ–Њ–ґ–µ—В —Б—В–∞—В—М –њ—А–Є—З–Є–љ–Њ–є –≤–љ–µ–Ј–∞–њ–љ–Њ–є —Б–Љ–µ—А—В–Є [4]. –Ю—В–ї–Њ–ґ–µ–љ–Є–µ –∞–Љ–Є–ї–Њ–Є–і–∞ —В–∞–Ї–ґ–µ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –Є –≤ —Б—В–µ–љ–Ї–∞—Е –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е —Б–Њ—Б—Г–і–Њ–≤, –≤—Л–Ј—Л–≤–∞—П –Є—И–µ–Љ–Є—О –Љ–Є–Њ–Ї–∞—А–і–∞. –Т–Њ–≤–ї–µ—З–µ–љ–Є–µ –Ї–ї–∞–њ–∞–љ–љ–Њ–≥–Њ –∞–њ–њ–∞—А–∞—В–∞ –≤—Б—В—А–µ—З–∞–µ—В—Б—П —Г 12–15% –±–Њ–ї—М–љ—Л—Е –Є –≤ 35% –њ—А–Є –∞–љ–∞—В–Њ–Љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е. –Ъ–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –Њ—В–Љ–µ—З–∞–µ—В—Б—П —Г—В–Њ–ї—Й–µ–љ–Є–µ, –і–µ—Д–Њ—А–Љ–∞—Ж–Є—П –Ї–ї–∞–њ–∞–љ–Њ–≤ —Б —А–∞–Ј–≤–Є—В–Є–µ–Љ –Є—Е –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є, –∞ –њ—А–Є –Њ—В–ї–Њ–ґ–µ–љ–Є–Є –≤ –Њ–±–ї–∞—Б—В–Є —Д–Є–±—А–Њ–Ј–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞ —Д–Њ—А–Љ–Є—А—Г–µ—В—Б—П —Б—В–µ–љ–Њ–Ј [5].
–Э–∞–Є–±–Њ–ї–µ–µ —В–Є–њ–Є—З–љ—Л–Љ–Є –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є, –≤—Л—П–≤–ї—П–µ–Љ—Л–Љ–Є –њ—А–Є –Є–љ—Б—В—А—Г–Љ–µ–љ—В–∞–ї—М–љ–Њ–Љ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є, —П–≤–ї—П—О—В—Б—П –љ–Є–Ј–Ї–Є–є –≤–Њ–ї—М—В–∞–ґ –≠–Ъ–У, –∞—А–Є—В–Љ–Є–Є –Є –љ–∞—А—Г—И–µ–љ–Є—П –њ—А–Њ–≤–Њ–і–Є–Љ–Њ—Б—В–Є, –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Ј—Г–±—Ж—Л Q, —Б–Є–Љ—Г–ї–Є—А—Г—О—Й–Є–µ –Є–љ—Д–∞—А–Ї—В –Љ–Є–Њ–Ї–∞—А–і–∞; –≠—Е–Њ–Ъ–У –њ—А–Є–Ј–љ–∞–Ї–Є: —Б–Є–Љ–Љ–µ—В—А–Є—З–љ–Њ–µ —Г—В–Њ–ї—Й–µ–љ–Є–µ —Б—В–µ–љ–Њ–Ї –ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤, –Ї–∞—А—В–Є–љ–∞ —А–µ—Б—В—А–Є–Ї—В–Є–≤–љ–Њ–є –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є–Є —Б –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є –і–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є; –Љ–љ–Њ–≥–Є–µ –∞–≤—В–Њ—А—Л –Њ–њ–Є—Б—Л–≤–∞—О—В —Н—Е–Њ–њ–Њ–Ј–Є—В–Є–≤–љ—Л–µ –≤–Ї–ї—О—З–µ–љ–Є—П –≤ –Љ–Є–Њ–Ї–∞—А–і–µ –≤ –≤–Є–і–µ –Љ–µ–ї–Ї–Є—Е –≥—А–∞–љ—Г–ї, –Ї–Њ—В–Њ—А—Л–µ —Б—З–Є—В–∞—О—В—Б—П —З–∞—Б—В–Є—Ж–∞–Љ–Є –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е –≤–Ї–ї—О—З–µ–љ–Є–є, –≤–Њ–Ј–Љ–Њ–ґ–µ–љ –њ–µ—А–Є–Ї–∞—А–і–Є–∞–ї—М–љ—Л–є –≤—Л–њ–Њ—В [6].
–Я–Њ—А–∞–ґ–µ–љ–Є–µ —А–µ—Б–њ–Є—А–∞—В–Њ—А–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –њ—А–Є —Б–Є—Б—В–µ–Љ–љ—Л—Е —Д–Њ—А–Љ–∞—Е –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –≤—Б—В—А–µ—З–∞–µ—В—Б—П –і–Њ—Б—В–∞—В–Њ—З–љ–Њ —З–∞—Б—В–Њ. –Ґ–∞–Ї, –њ–Њ –і–∞–љ–љ—Л–Љ –Ю.–Ь. –Т–Є–љ–Њ–≥—А–∞–і–Њ–≤–Њ–є (1980), –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П —А–µ—Б–њ–Є—А–∞—В–Њ—А–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л –љ–∞–±–ї—О–і–∞–ї–Є—Б—М —Г 50% –±–Њ–ї—М–љ—Л—Е –Є —Г 83% –њ–Њ –њ–∞—В–Њ–ї–Њ–≥–Њ–∞–љ–∞—В–Њ–Љ–Є—З–µ—Б–Ї–Є–Љ –і–∞–љ–љ—Л–Љ.
–Т—Л–і–µ–ї—П—О—В –і–Є—Д—Д—Г–Ј–љ–Њ–µ –Є –ї–Њ–Ї–∞–ї—М–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е. Weis (1960) –≤ —Б–≤–Њ–µ–є –Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є–Є –≤—Л–і–µ–ї—П–ї –≥–µ–љ–µ—А–∞–ї–Є–Ј–Њ–≤–∞–љ–љ—Л–є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј, –њ–Њ–і—А–∞–Ј–і–µ–ї—П–µ–Љ—Л–є –љ–∞ –љ–Њ–і—Г–ї—П—А–љ—Л–є –Є –і–Є—Д—Д—Г–Ј–љ—Л–є –ї–µ–≥–Њ—З–љ—Л–є –Є –Њ–≥—А–∞–љ–Є—З–µ–љ–љ—Л–є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј, –≤ –Ї–Њ—В–Њ—А–Њ–Љ –Њ—В–і–µ–ї—М–љ–Њ –≤—Л–і–µ–ї—П–ї—Б—П —В—А–∞—Е–µ–Њ–±—А–Њ–љ—Е–Є–∞–ї—М–љ—Л–є –Є –Є–Ј–Њ–ї–Є—А–Њ–≤–∞–љ–љ—Л–є –љ–Њ–і—Г–ї—П—А–љ—Л–є –ї–µ–≥–Њ—З–љ—Л–є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј. –Т–њ–Њ—Б–ї–µ–і—Б—В–≤–Є–Є –±—Л–ї–Є –Њ–њ–Є—Б–∞–љ—Л —Б–Љ–µ—И–∞–љ–љ—Л–µ –і–Є—Д—Д—Г–Ј–љ–Њ––љ–Њ–і—Г–ї—П—А–љ—Л–µ —Д–Њ—А–Љ—Л –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞). John L. Berk –≤—Л–і–µ–ї—П–µ—В 5 —Д–Њ—А–Љ —Н–Ї—Б—В—А–∞–≤–∞—Б–Ї—Г–ї—П—А–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Ї–Є—Е –њ—А–Є –РL––∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ [7]:
1. –Ф–Є—Д—Д—Г–Ј–љ–Њ––Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–µ, –Є–ї–Є –∞–ї—М–≤–µ–Њ–ї—П—А–љ–Њ–—Б–µ–њ—В–∞–ї—М–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–Љ –∞–Љ–Є–ї–Њ–Є–і –Њ—В–Ї–ї–∞–і—Л–≤–∞–µ—В—Б—П –Љ–µ–ґ–і—Г —Н–љ–і–Њ—В–µ–ї–Є–µ–Љ —Б–Њ—Б—Г–і–Њ–≤ –Є –∞–ї—М–≤–µ–Њ–ї—П—А–љ—Л–Љ —Н–њ–Є—В–µ–ї–Є–µ–Љ –ї–µ–≥–Њ—З–љ–Њ–≥–Њ –Є–љ—В–µ—А—Б—В–Є—Ж–Є—П.
2. –Э–Њ–і—Г–ї—П—А–љ–Њ–µ –Њ—В–ї–Њ–ґ–µ–љ–Є–µ.
3. –Я–Њ—А–∞–ґ–µ–љ–Є–µ –њ–ї–µ–≤—А—Л.
4. –Ш–љ—В—А–∞– –Є —Н–Ї—Б—В—А–∞—В–Њ—А–∞–Ї–∞–ї—М–љ–∞—П –∞–і–µ–љ–Њ–њ–∞—В–Є—П.
5. –Я–Њ—А–∞–ґ–µ–љ–Є–µ –і–Є–∞—Д—А–∞–≥–Љ—Л, –≤—Б—В—А–µ—З–∞—О—Й–µ–µ—Б—П –Ї—А–∞–є–љ–µ —А–µ–і–Ї–Њ.
–Ю—В–і–µ–ї—М–љ–Њ–≥–Њ –≤–љ–Є–Љ–∞–љ–Є—П –Ј–∞—Б–ї—Г–ґ–Є–≤–∞–µ—В –і–Є—Д—Д—Г–Ј–љ–Њ––Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–∞—П —Д–Њ—А–Љ–∞, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –Є–Љ–µ–љ–љ–Њ –Њ–љ–∞ —Б–Њ—З–µ—В–∞–µ—В—Б—П —Б –Ї–∞—А–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–Є–Љ –≤–∞—А–Є–∞–љ—В–Њ–Љ —В–µ—З–µ–љ–Є—П –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ –Є —З–∞—Б—В–Њ –љ–µ –і–Є–∞–≥–љ–Њ—Б—В–Є—А—Г–µ—В—Б—П, —В–∞–Ї –Ї–∞–Ї –µ–µ –Ї–ї–Є–љ–Є–Ї–Њ–—А–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –њ—А–Є–љ–Є–Љ–∞–µ—В—Б—П –Ј–∞ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Ј–∞—Б—В–Њ–є–љ–Њ–є —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є. John L. Berk [7] –њ—А–µ–і—Б—В–∞–≤–ї—П–ї –і–∞–љ–љ—Л–µ –∞—Г—В–Њ–њ—Б–Є–Є 12 –РL––њ–∞—Ж–Є–µ–љ—В–Њ–≤: —Г –≤—Б–µ—Е –Њ–њ—А–µ–і–µ–ї—П–ї–Є—Б—М –і–µ–њ–Њ–Ј–Є—В—Л –≤ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–Є, –∞–ї—М–≤–µ–Њ–ї—П—А–љ—Л—Е –њ–µ—А–µ–≥–Њ—А–Њ–і–Ї–∞—Е, –љ–∞ –±–∞–Ј–∞–ї—М–љ—Л—Е –Љ–µ–Љ–±—А–∞–љ–∞—Е –Љ–µ–ґ–і—Г –Ї–ї–µ—В–Ї–∞–Љ–Є –∞–ї—М–≤–µ–Њ–ї—П—А–љ–Њ–≥–Њ —Н–њ–Є—В–µ–ї–Є—П –Є –Ї–ї–µ—В–Ї–∞–Љ–Є —Н–љ–і–Њ—В–µ–ї–Є—П –Ї–∞–њ–Є–ї–ї—П—А–Њ–≤, —Б—В–µ–љ–Ї–∞—Е –Љ–µ–ї–Ї–Є—Е —Б–Њ—Б—Г–і–Њ–≤ –Є –≤–Њ–Ј–і—Г—И–љ—Л—Е –њ—Г—В—П—Е. –Э–µ—Б–Љ–Њ—В—А—П –љ–∞ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ—Л–µ –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П, —Г 64% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Њ—В—Б—Г—В—Б—В–≤–Њ–≤–∞–ї–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П –і—Л—Е–∞—В–µ–ї—М–љ—Л—Е –њ—Г—В–µ–є. –Ґ–Њ–ї—М–Ї–Њ –≤ 1 —Б–ї—Г—З–∞–µ –Є–Ј 12 –њ–Њ—А–∞–ґ–µ–љ–Є–µ –ї–µ–≥–Ї–Є—Е –њ—А–µ–і–Њ–њ—А–µ–і–µ–ї–Є–ї–Њ –Є—Б—Е–Њ–і –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Я–Њ –і–∞–љ–љ—Л–Љ –і—А—Г–≥–Є—Е –∞–≤—В–Њ—А–Њ–≤, –і–Є—Д—Д—Г–Ј–љ–Њ––Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ —П–≤–Є–ї–Њ—Б—М –њ—А–Є—З–Є–љ–Њ–є —Б–Љ–µ—А—В–Є 10% –њ–∞—Ж–Є–µ–љ—В–Њ–≤.
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ–Є –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є —Н—В–Њ–≥–Њ –≤–∞—А–Є–∞–љ—В–∞ —П–≤–ї—П—О—В—Б—П –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–∞—П –Њ–і—Л—И–Ї–∞ –Є —Б—Г—Е–Њ–є –њ—А–Є—Б—В—Г–њ–Њ–Њ–±—А–∞–Ј–љ—Л–є –Ї–∞—И–µ–ї—М. –Я—А–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Д—Г–љ–Ї—Ж–Є–Є –≤–љ–µ—И–љ–µ–≥–Њ –і—Л—Е–∞–љ–Є—П –≤—Л—П–≤–ї—П–µ—В—Б—П —А–µ—Б—В—А–Є–Ї—В–Є–≤–љ—Л–є —В–Є–њ –≤–µ–љ—В–Є–ї—П—Ж–Є–Њ–љ–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є. –Я—А–Є —А–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ –Є –Ъ–Ґ––Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –Њ—В–Љ–µ—З–∞—О—В –і–Є—Д—Д—Г–Ј–љ–Њ–µ —Г—Б–Є–ї–µ–љ–Є–µ –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —А–Є—Б—Г–љ–Ї–∞ –Ј–∞ —Б—З–µ—В —Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–∞.
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –њ–ї–µ–≤—А—Л –њ—А–Є AL––∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ –≤—Б—В—А–µ—З–∞–µ—В—Б—П —А–µ–і–Ї–Њ –Є –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–Є–±–ї–Є–Ј–Є—В–µ–ї—М–љ–Њ —Г 6% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ (–њ–Њ –і–∞–љ–љ—Л–Љ –Ю.–Ь. –Т–Є–љ–Њ–≥—А–∞–і–Њ–≤–Њ–є, —Б–Њ—Б—В–∞–≤–ї—П–µ—В 6,6%). John L. Berk —Г–Ї–∞–Ј—Л–≤–∞–µ—В 6% [8], —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ–Љ –њ–ї–µ–≤—А–Є—В–∞ –∞–≥—А–µ—Б—Б–Є–≤–љ–Њ–≥–Њ —В–µ—З–µ–љ–Є—П –Є —В–Њ—А–њ–Є–і–љ–Њ–≥–Њ –Ї –њ—А–Њ–≤–Њ–і–Є–Љ–Њ–є –і–Є—Г—А–µ—В–Є—З–µ—Б–Ї–Њ–є —В–µ—А–∞–њ–Є–Є. –Я–ї–µ–≤—А–∞–ї—М–љ—Л–є –≤—Л–њ–Њ—В –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ–і–љ–Њ—Б—В–Њ—А–Њ–љ–љ–Є–Љ, –љ–Њ —З–∞—Й–µ – –±–Є–ї–∞—В–µ—А–∞–ї—М–љ—Л–є. –Я—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –љ–Њ—Б–Є—В —В—А–∞–љ—Б—Б—Г–і–∞—В–Є–≤–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А, —Е–Њ—В—П –±—Л–≤–∞—О—В –Є —Н–Ї—Б—Б—Г–і–∞—В—Л. –Т —Ж–Є—В–Њ–≥—А–∞–Љ–Љ–µ –њ—А–µ–Њ–±–ї–∞–і–∞–µ—В –ї–Є–Љ—Д–Њ—Ж–Є—В–Њ–Ј. –Я–ї–µ–≤—А–∞–ї—М–љ—Л–є –≤—Л–њ–Њ—В –≤–µ—Б—М–Љ–∞ —З–∞—Б—В–Њ –≤—Б—В—А–µ—З–∞–µ—В—Б—П —Г –±–Њ–ї—М–љ—Л—Е —Б –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є–µ–є, –њ–Њ—Н—В–Њ–Љ—Г —А–∞–Ј–≤–Є—В–Є–µ –њ–ї–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –≤—Л–њ–Њ—В–∞ —Б–≤—П–Ј—Л–≤–∞–µ—В—Б—П —Б –і–µ–Ї–Њ–Љ–њ–µ–љ—Б–Є—А–Њ–≤–∞–љ–љ–Њ–є —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О. –Ю–і–љ–∞–Ї–Њ –њ—А–Њ–≤–µ–і–µ–љ–љ–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ —Ж–µ–љ—В—А–∞ –С–Њ—Б—В–Њ–љ—Б–Ї–Њ–≥–Њ —Г–љ–Є–≤–µ—А—Б–Є—В–µ—В–∞, –≤–Ї–ї—О—З–∞–≤—И–µ–µ 636 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Ј–∞ –њ–µ—А–Є–Њ–і 1994–2001 –≥–≥., –Њ–њ—А–Њ–≤–µ—А–≥–∞–µ—В —Н—В–Њ –Љ–љ–µ–љ–Є–µ [8]. –Р–љ–∞–ї–Є–Ј–Є—А–Њ–≤–∞–ї–Є—Б—М –і–≤–µ –≥—А—Г–њ–њ—Л –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –РL––∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ: –≤ –њ–µ—А–≤—Г—О –≤—Е–Њ–і–Є–ї–Є –њ–∞—Ж–Є–µ–љ—В—Л —Б –∞–Љ–Є–ї–Њ–Є–і–љ—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —Б–µ—А–і—Ж–∞ –Є –њ–ї–µ–≤—А–∞–ї—М–љ—Л–Љ –≤—Л–њ–Њ—В–Њ–Љ (35 —З–µ–ї.), –≤—В–Њ—А–∞—П –≥—А—Г–њ–њ–∞ —Б –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є–µ–є –±–µ–Ј –њ–ї–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –≤—Л–њ–Њ—В–∞ (120 —З–µ–ї.) [8]. –†–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –Є –љ–Є–Ј–Ї–Њ–µ –Њ–љ–Ї–Њ—В–Є—З–µ—Б–Ї–Њ–µ –і–∞–≤–ї–µ–љ–Є–µ –њ–ї–∞–Ј–Љ—Л –љ–µ —П–≤–ї—П—О—В—Б—П –Њ—Б–љ–Њ–≤–Њ–њ–Њ–ї–∞–≥–∞—О—Й–Є–Љ–Є –≤ —А–∞–Ј–≤–Є—В–Є–Є –њ–ї–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –≤—Л–њ–Њ—В–∞. –Ю–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –њ–ї–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –≤—Л–њ–Њ—В–∞ —Б–≤—П–Ј–∞–љ–Њ –≤ –њ–µ—А–≤—Г—О –Њ—З–µ—А–µ–і—М —Б –Њ—В–ї–Њ–ґ–µ–љ–Є–µ–Љ –∞–Љ–Є–ї–Њ–Є–і–∞ –≤ –Љ–µ–ґ–Ї–ї–µ—В–Њ—З–љ—Л—Е –њ—А–Њ–Љ–µ–ґ—Г—В–Ї–∞—Е, –≤ —Б—Г–±–Љ–µ–Ј–Њ—В–µ–ї–Є–∞–ї—М–љ—Л—Е –ї–Є–Љ—Д–∞—В–Є—З–µ—Б–Ї–Є—Е —Б–Њ—Б—Г–і–∞—Е. –Р–Љ–Є–ї–Њ–Є–і–љ—Л–µ –і–µ–њ–Њ–Ј–Є—В—Л –Є–љ–≥–Є–±–Є—А—Г—О—В —А–µ–Ј–Њ—А–±—Ж–Є—О –њ–ї–µ–≤—А–∞–ї—М–љ–Њ–є –ґ–Є–і–Ї–Њ—Б—В–Є, –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —З–µ–≥–Њ —Б—В—А–∞–і–∞–µ—В –і—А–µ–љ–∞–ґ–љ–∞—П —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –њ–ї–µ–≤—А—Л. –Т —Б–≤–Њ—О –Њ—З–µ—А–µ–і—М, –∞–Љ–Є–ї–Њ–Є–і–љ—Л–µ –Њ—В–ї–Њ–ґ–µ–љ–Є—П –Є–Ј–Љ–µ–љ—П—О—В –Є —Б–µ–Ї—А–µ—В–Њ—А–љ—Г—О —Д—Г–љ–Ї—Ж–Є—О –њ–ї–µ–≤—А—Л [9].
–Ф–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–ї–Њ–ґ–љ–∞ –Є —В—А–µ–±—Г–µ—В –њ—А–Њ–≤–µ–і–µ–љ–Є—П –Ї–Њ–Љ–њ–ї–µ–Ї—Б–∞ –ї–∞–±–Њ—А–∞—В–Њ—А–љ–Њ––Є–љ—Б—В—А—Г–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –љ–Њ –Њ–Ї–Њ–љ—З–∞—В–µ–ї—М–љ–Њ –і–Є–∞–≥–љ–Њ–Ј –і–Њ–ї–ґ–µ–љ –±—Л—В—М –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є. –Ш–Ј–Љ–µ–љ–µ–љ–Є—П –ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л—Е –і–∞–љ–љ—Л—Е –љ–µ—Б–њ–µ—Ж–Є—Д–Є—З–љ—Л –Є –Њ—В—А–∞–ґ–∞—О—В –љ–∞—А—Г—И–µ–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–Є —В–Њ–≥–Њ –Є–ї–Є –Є–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–љ–Њ–≥–Њ –Њ—А–≥–∞–љ–∞. –£ 85% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –њ—А–Є –Є–Љ–Љ—Г–љ–Њ—Н–ї–µ–Ї—В—А–Њ—Д–Њ—А–µ—В–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Б—Л–≤–Њ—А–Њ—В–Ї–Є –±–µ–ї–Ї–∞ –Є –Љ–Њ—З–Є –≤—Л—П–≤–ї—П–µ—В—Б—П –Љ–Њ–љ–Њ–Ї–ї–Њ–љ–∞–ї—М–љ—Л–є –њ–∞—А–∞–њ—Аo—В–µ–Є–љ, –Ї–Њ—В–Њ—А—Л–є –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ –ї–µ–≥–Ї–Є–Љ–Є —Ж–µ–њ—П–Љ–Є –Є–Љ–Љ—Г–љ–Њ–≥–ї–Њ–±—Г–ї–Є–љ–Њ–≤ —З–∞—Й–µ –ї–, —А–µ–ґ–µ –Ї–—Ж–µ–њ–µ–є. –Я—А–Є –Ї–Њ–∞–≥—Г–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –≤—Л—П–≤–ї—П–µ—В—Б—П –і–µ—Д–Є—Ж–Є—В –• —Д–∞–Ї—В–Њ—А–∞ —Б–≤–µ—А—В—Л–≤–∞–љ–Є—П. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Ї–Њ—Б—В–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –њ–Њ–Ї–∞–Ј—Л–≤–∞–µ—В –µ–≥–Њ —Г–Љ–µ—А–µ–љ–љ—Г—О –њ–ї–∞–Ј–Љ–∞—В–Є–Ј–∞—Ж–Є—О 5–10% (–і–Њ 20%). –°–Њ—З–µ—В–∞–љ–Є–µ —Е–∞—А–∞–Ї—В–µ—А–љ–Њ–є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ—Л —Б –љ–∞–ї–Є—З–Є–µ–Љ –Љ–Њ–љ–Њ–Ї–ї–Њ–љ–∞–ї—М–љ—Л—Е –±–µ–ї–Ї–Њ–≤ –≤ —Б—Л–≤–Њ—А–Њ—В–Ї–µ –Ї—А–Њ–≤–Є –Є –Љ–Њ—З–Є –Є —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї –≤ –Ї–Њ—Б—В–љ–Њ–Љ –Љ–Њ–Ј–≥–µ —П–≤–ї—П–µ—В—Б—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ—Л–Љ –і–ї—П —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –і–Є–∞–≥–љ–Њ–Ј–∞ –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Ф–Њ—Б—В–Њ–≤–µ—А–љ—Л–Љ –Љ–µ—В–Њ–і–Њ–Љ —П–≤–ї—П–µ—В—Б—П –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–Њ—А–∞–ґ–µ–љ–љ–Њ–≥–Њ –Њ—А–≥–∞–љ–∞ (–њ–Њ—З–Ї–∞, –њ–µ—З–µ–љ—М, –Љ–Є–Њ–Ї–∞—А–і –Є –і—А.) [10]. –Ф–Њ—Б—В–∞—В–Њ—З–љ–Њ –Є–љ—Д–Њ—А–Љ–∞—В–Є–≤–љ–Њ–є —П–≤–ї—П–µ—В—Б—П –±–Є–Њ–њ—Б–Є—П —Б–ї–Є–Ј–Є—Б—В–Њ–є –њ—А—П–Љ–Њ–є –Ї–Є—И–Ї–Є, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–є –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М –≤—Л—П–≤–ї–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞ —Б–Њ—Б—В–∞–≤–ї—П–µ—В 50–70%. –С–Є–Њ–њ—Б–Є—П —Б–ї–Є–Ј–Є—Б—В–Њ–є –і–µ—Б–љ—Л –Љ–∞–ї–Њ–Є–љ—Д–Њ—А–Љ–∞—В–Є–≤–љ–∞, –Ј–∞ —А—Г–±–µ–ґ–Њ–Љ —И–Є—А–Њ–Ї–Њ –Є—Б–њ–Њ–ї—М–Ј—Г—О—В –∞—Б–њ–Є—А–∞—Ж–Є–Њ–љ–љ—Г—О –±–Є–Њ–њ—Б–Є—О –њ–Њ–і–Ї–Њ–ґ–љ–Њ––ґ–Є—А–Њ–≤–Њ–є –Ї–ї–µ—В—З–∞—В–Ї–Є –Є–Ј –њ–µ—А–µ–і–љ–µ–є –±—А—О—И–љ–Њ–є —Б—В–µ–љ–Ї–Є. –І–µ–Љ –±–Њ–ї–µ–µ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ –њ—А–Њ—Ж–µ—Б—Б, —В–µ–Љ –≤—Л—И–µ –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М –≤—Л—П–≤–ї–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞ –≤ —А–∞–Ј–ї–Є—З–љ—Л—Е –Љ–µ—Б—В–∞—Е. –Ф–ї—П –Њ–њ—А–µ–і–µ–ї–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞ –≤ –≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ –Љ–∞—В–µ—А–Є–∞–ї–µ –Є—Б–њ–Њ–ї—М–Ј—Г—О—В –Њ–Ї—А–∞—Б–Њ—З–љ—Л–µ –Љ–µ—В–Њ–і—Л: –њ—А–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ–Є–Є –Ї–Њ–љ–≥–Њ—Д–Є–ї–Є–Є –њ—А–µ–њ–∞—А–∞—В –Є—Б—Б–ї–µ–і—Г—О—В –≤ –њ–Њ–ї—П—А–Є–Ј–Њ–≤–∞–љ–љ–Њ–Љ —Б–≤–µ—В–µ –і–ї—П –≤—Л—П–≤–ї–µ–љ–Є—П —Н—Д—Д–µ–Ї—В–∞ –і–≤–Њ–є–љ–Њ–≥–Њ –ї—Г—З–µ–њ—А–µ–ї–Њ–Љ–ї–µ–љ–Є—П, –њ—А–Є —Н—В–Њ–Љ —В–Њ–ї—М–Ї–Њ –∞–Љ–Є–ї–Њ–Є–і –њ—А–Є–Њ–±—А–µ—В–∞–µ—В —П–±–ї–Њ—З–љ–Њ––Ј–µ–ї–µ–љ—Г—О –Њ–Ї—А–∞—Б–Ї—Г. –Ф–ї—П —В–Є–њ–Є—А–Њ–≤–∞–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–∞ –њ–Њ–ї—М–Ј—Г—О—В—Б—П –Њ–Ї—А–∞—Б–Њ—З–љ—Л–Љ –Љ–µ—В–Њ–і–Њ–Љ —Б —А–∞—Б—В–≤–Њ—А–Њ–Љ —Й–µ–ї–Њ—З–љ–Њ–≥–Њ –≥—Г–∞–љ–Є–і–Є–љ–∞, –Ї–Њ—В–Њ—А—Л–є –Є–Ј–Љ–µ–љ—П–µ—В –Ї–Њ–љ–≥–Њ—Д–Є–ї—М–љ—Л–µ —Б–≤–Њ–є—Б—В–≤–∞ –∞–Љ–Є–ї–Њ–Є–і–∞ —А–∞–Ј–ї–Є—З–љ—Л—Е —В–Є–њ–Њ–≤ –≤ –Ј–∞–≤–Є—Б–Є–Љ–Њ—Б—В–Є –Њ—В –≤—А–µ–Љ–µ–љ–Є —Н–Ї—Б–њ–Њ–Ј–Є—Ж–Є–Є –≤ —А–∞—Б—В–≤–Њ—А–µ –≥—Г–∞–љ–Є–і–Є–љ–∞. –Э–Њ –љ–∞–Є–±–Њ–ї–µ–µ —В–Њ—З–љ—Л–Љ —П–≤–ї—П–µ—В—Б—П –Є–Љ–Љ—Г–љ–Њ–≥–Є—Б—В–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ —Б –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ –Љ–Њ–љ–Њ–Ї–ї–Њ–љ–∞–ї—М–љ—Л—Е –∞–љ—В–Є—В–µ–ї –Ї –±–µ–ї–Ї–∞–Љ – –њ—А–µ–і—И–µ—Б—В–≤–µ–љ–љ–Є–Ї–∞–Љ –∞–Љ–Є–ї–Њ–Є–і–∞.
–Ф–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —П–≤–ї—П–µ—В—Б—П —В—А—Г–і–љ–Њ–є –Ј–∞–і–∞—З–µ–є, —З—В–Њ –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ–Њ–µ –љ–∞–Љ–Є –љ–∞–±–ї—О–і–µ–љ–Є–µ.
–С–Њ–ї—М–љ–∞—П –Ъ., 54 –ї–µ—В, –њ–Њ—Б—В—Г–њ–Є–ї–∞ –≤ –Ї–ї–Є–љ–Є–Ї—Г —Б –ґ–∞–ї–Њ–±–∞–Љ–Є –љ–∞ –Њ–і—Л—И–Ї—Г –њ—А–Є –љ–µ–±–Њ–ї—М—И–Њ–є —Д–Є–Ј–Є—З–µ—Б–Ї–Њ–є –љ–∞–≥—А—Г–Ј–Ї–µ, —Б—Г—Е–Њ–є –Ї–∞—И–µ–ї—М, –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–Є–є –њ—А–Є –њ–µ—А–µ–Љ–µ–љ–µ –њ–Њ–ї–Њ–ґ–µ–љ–Є—П —В–µ–ї–∞, –і–∞–≤—П—Й–Є–µ –±–Њ–ї–Є –≤ –≥—А—Г–і–Є –њ—А–Є —Д–Є–Ј–Є—З–µ—Б–Ї–Њ–є –љ–∞–≥—А—Г–Ј–Ї–µ –Є –≤ –њ–Њ–Ї–Њ–µ, –≤—Л—А–∞–ґ–µ–љ–љ—Г—О –Њ–±—Й—Г—О —Б–ї–∞–±–Њ—Б—В—М, –Њ—В—Б—Г—В—Б—В–≤–Є–µ –∞–њ–њ–µ—В–Є—В–∞, —Б–љ–Є–ґ–µ–љ–Є–µ –≤–µ—Б–∞ –Ј–∞ –њ–Њ—Б–ї–µ–і–љ–Є–є –≥–Њ–і –љ–∞ 16 –Ї–≥.
–Ш–Ј –∞–љ–∞–Љ–љ–µ–Ј–∞ –Є–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –≤–њ–µ—А–≤—Л–µ —Б—В–∞–ї–∞ –Њ—В–Љ–µ—З–∞—В—М –њ–Њ–≤—Л—И–µ–љ–Є–µ –Р–Ф –≤–Њ –≤—А–µ–Љ—П –њ–µ—А–≤–Њ–є –±–µ—А–µ–Љ–µ–љ–љ–Њ—Б—В–Є –≤ 23 –≥–Њ–і–∞, –і–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞–љ–∞ –љ–µ—Д—А–Њ–њ–∞—В–Є—П –±–µ—А–µ–Љ–µ–љ–љ–Њ—Б—В–Є. –° 30––ї–µ—В–љ–µ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞ —Г—А–Њ–≤–µ–љ—М –Р–Ф —Б—В–∞–±–Є–ї–Є–Ј–Є—А–Њ–≤–∞–ї—Б—П –љ–∞ —Ж–Є—Д—А–∞—Е 140–150/90–100 –Љ–Љ —А—В.—Б—В. —Б –њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ –і–Њ 170–190/90–100 –Љ–Љ —А—В.—Б—В. –Я–Њ—Б—В–Њ—П–љ–љ—Г—О –≥–Є–њ–Њ—В–µ–љ–Ј–Є–≤–љ—Г—О —В–µ—А–∞–њ–Є—О –љ–µ –њ–Њ–ї—Г—З–∞–ї–∞, —Б–Є—В—Г–∞—Ж–Є–Њ–љ–љ–Њ –њ—А–Є–љ–Є–Љ–∞–ї–∞ –Ї–ї–Њ–љ–Є–і–Є–љ. –Т 1986 –≥. –њ—А–Њ–≤–Њ–і–Є–ї–∞—Б—М —Б—Ж–Є–љ—В–Є–≥—А–∞—Д–Є—П –њ–Њ—З–µ–Ї, –≤—Л—П–≤–Є–≤—И–∞—П –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –≤—Л–і–µ–ї–Є—В–µ–ї—М–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є —Б –Њ–±–µ–Є—Е —Б—В–Њ—А–Њ–љ, –±–µ–Ј –Є–Ј–Љ–µ–љ–µ–љ–Є—П —Б–µ–Ї—А–µ—В–Њ—А–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є. –£–Ч––Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –≤—Л—П–≤–Є–ї–Њ –і–≤—Г—Б—В–Њ—А–Њ–љ–љ—О—О –њ–Є–µ–ї–Њ—Н–Ї—В–∞–Ј–Є—О. –Т –∞–љ–∞–ї–Є–Ј–∞—Е –Љ–Њ—З–Є –љ–∞–±–ї—О–і–∞–ї–∞—Б—М –ї–µ–є–Ї–Њ—Ж–Є—В—Г—А–Є—П. –Э–∞ –Њ—Б–љ–Њ–≤–∞–љ–Є–Є —Н—В–Њ–≥–Њ –±—Л–ї–Њ —Б–і–µ–ї–∞–љ–Њ –Ј–∞–Ї–ї—О—З–µ–љ–Є–µ –Њ –љ–∞–ї–Є—З–Є–Є —Г –±–Њ–ї—М–љ–Њ–є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Є–µ–ї–Њ–љ–µ—Д—А–Є—В–∞ –Є —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–Є –≤—В–Њ—А–Є—З–љ–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є.
–° 1998 –≥–Њ–і–∞ –±–Њ–ї—М–љ–∞—П —Б—В–∞–ї–∞ –Њ—В–Љ–µ—З–∞—В—М –њ–Њ—П–≤–ї–µ–љ–Є–µ –≥–µ–Љ–Њ—А—А–∞–≥–Є—З–µ—Б–Ї–Є—Е –≤—Л—Б—Л–њ–∞–љ–Є–є —А–∞–Ј–Љ–µ—А–Њ–Љ —Б–Њ —Б–њ–Є—З–µ—З–љ—Г—О –≥–Њ–ї–Њ–≤–Ї—Г –љ–∞ –Ї–Њ–ґ–µ –њ–∞—А–∞–Њ—А–±–Є—В–∞–ї—М–љ—Л—Е –Њ–±–ї–∞—Б—В–µ–є, –Ї–Њ—В–Њ—А—Л–µ —Б–Њ –≤—А–µ–Љ–µ–љ–µ–Љ –≥–Є–њ–µ—А–њ–Є–≥–Љ–µ–љ—В–Є—А–Њ–≤–∞–ї–Є—Б—М. –Т 2000 –≥–Њ–і—Г –љ–∞ —Д–Њ–љ–µ –Ю–†–Т–Ш –њ–Њ—П–≤–ї—П–µ—В—Б—П —Б—В–Њ–є–Ї–∞—П –Њ—Б–Є–њ–ї–Њ—Б—В—М –≥–Њ–ї–Њ—Б–∞. –Т 2001 –њ–Њ—Б–ї–µ –њ–µ—А–µ–љ–µ—Б–µ–љ–љ–Њ–є –љ–∞–і–≤–ї–∞–≥–∞–ї–Є—Й–љ–Њ–є –∞–Љ–њ—Г—В–∞—Ж–Є–Є –Љ–∞—В–Ї–Є —Б –ї–µ–≤—Л–Љ–Є –њ—А–Є–і–∞—В–Ї–∞–Љ–Є –њ–Њ –њ–Њ–≤–Њ–і—Г –Љ–Є–Њ–Љ—Л –Љ–∞—В–Ї–Є –Є —Н–љ–і–Њ–Љ–µ—В—А–Є–Њ–Ј–∞ –ї–µ–≤–Њ–≥–Њ —П–Є—З–љ–Є–Ї–∞, —В–µ—З–µ–љ–Є–µ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є –Њ—Б–ї–Њ–ґ–љ–Є–ї–Њ—Б—М –≤—Л—Б–Њ–Ї–Є–Љ–Є —Ж–Є—Д—А–∞–Љ–Є, –њ–∞—Ж–Є–µ–љ—В–Ї–∞ —Б—В–∞–ї–∞ –Њ—В–Љ–µ—З–∞—В—М –њ–Њ—П–≤–ї–µ–љ–Є–µ –Њ–і—Л—И–Ї–Є –њ—А–Є –Њ–±—Л—З–љ—Л—Е —Д–Є–Ј–Є—З–µ—Б–Ї–Є—Е –љ–∞–≥—А—Г–Ј–Ї–∞—Е. –Я—А–Є –≠—Е–Њ–Ъ–У –≤–њ–µ—А–≤—Л–µ –≤—Л—П–≤–ї–µ–љ–∞ –≥–Є–њ–µ—А—В—А–Њ—Д–Є—П –Љ–Є–Њ–Ї–∞—А–і–∞ –Ы–Ц (—В–Њ–ї—Й–Є–љ–∞ –Љ–µ–ґ–ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤–Њ–є –њ–µ—А–µ–≥–Њ—А–Њ–і–Ї–Є –Є —В–Њ–ї—Й–Є–љ–∞ –Ј–∞–і–љ–µ–є —Б—В–µ–љ–Ї–Є —Б–Њ—Б—В–∞–≤–ї—П–ї–∞ 12 –Љ–Љ) –±–µ–Ј –і–Є–ї–∞—В–∞—Ж–Є–Є –Ї–∞–Љ–µ—А —Б–µ—А–і—Ж–∞. –С—Л–ї –љ–∞–Ј–љ–∞—З–µ–љ —Н–љ–∞–ї–∞–њ—А–Є–ї, –Ї–Њ—В–Њ—А—Л–є –њ—А–Є–љ–Є–Љ–∞–ї–∞ –љ–µ—А–µ–≥—Г–ї—П—А–љ–Њ. –Я–Њ—Б–ї–µ —Б—В—А–µ—Б—Б–∞ –≤ —П–љ–≤–∞—А–µ 2003 –≥. –њ–Њ—П–≤–Є–ї–Є—Б—М –љ–µ–Є–љ—В–µ–љ—Б–Є–≤–љ—Л–µ –і–∞–≤—П—Й–Є–µ –±–Њ–ї–Є –Ј–∞ –≥—А—Г–і–Є–љ–Њ–є, —Б–љ–Є–ґ–µ–љ–Є–µ —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є –Ї —Д–Є–Ј–Є—З–µ—Б–Ї–Є–Љ –љ–∞–≥—А—Г–Ј–Ї–∞–Љ –≤ –≤–Є–і–µ –Њ–і—Л—И–Ї–Є, —Б–ї–∞–±–Њ—Б—В—М. –Ъ –≤—А–∞—З–∞–Љ –љ–µ –Њ–±—А–∞—Й–∞–ї–∞—Б—М. –Ы–µ—В–Њ–Љ –Њ—В–Љ–µ—В–Є–ї–∞ –њ–Њ—П–≤–ї–µ–љ–Є–µ –Њ—В–µ–Ї–Њ–≤ –љ–∞ –љ–Є–ґ–љ–Є—Е –Ї–Њ–љ–µ—З–љ–Њ—Б—В—П—Е –Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –Њ–і—Л—И–Ї–Є. –Т —Б–µ–љ—В—П–±—А–µ –њ–Њ—Б–ї–µ —Д–Є–Ј–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–µ—А–µ–љ–∞–њ—А—П–ґ–µ–љ–Є—П –≤ —В–µ—З–µ–љ–Є–µ –љ–µ–і–µ–ї–Є –Њ—В–Љ–µ—В–Є–ї–∞ —А–µ–Ј–Ї–Њ–µ —Г—Е—Г–і—И–µ–љ–Є–µ —Б–∞–Љ–Њ—З—Г–≤—Б—В–≤–Є—П: —Г—Б–Є–ї–µ–љ–Є–µ –Њ–і—Л—И–Ї–Є, –њ–Њ—П–≤–ї–µ–љ–Є–µ –Є–љ—В–µ–љ—Б–Є–≤–љ—Л—Е –±–Њ–ї–µ–є –Ј–∞ –≥—А—Г–і–Є–љ–Њ–є, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –Њ—В–µ–Ї–Њ–≤ –≥–Њ–ї–µ–љ–µ–є –Є —Б—В–Њ–њ. –Я—А–Є –Њ–±—А–∞—Й–µ–љ–Є–Є –≤ –њ–Њ–ї–Є–Ї–ї–Є–љ–Є–Ї—Г –љ–∞ –≠–Ъ–У –≤—Л—П–≤–ї–µ–љ—Л —А—Г–±—Ж–Њ–≤—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ–µ—А–µ–і–љ–µ––њ–µ—А–µ–≥–Њ—А–Њ–і–Њ—З–љ–Њ–є –Њ–±–ї–∞—Б—В–Є –Є –≤–µ—А—Е—Г—И–Ї–Є –Ы–Ц, –њ—А–Є –≠—Е–Њ–Ъ–У – –∞–Ї–Є–љ–µ–Ј –њ–µ—А–µ–і–љ–µ–—Б–µ–њ—В–∞–ї—М–љ–Њ–є –Њ–±–ї–∞—Б—В–Є –Є —Б–љ–Є–ґ–µ–љ–Є–µ —Д—А–∞–Ї—Ж–Є–Є –≤—Л–±—А–Њ—Б–∞ –Ы–Ц; –њ–Њ –і–∞–љ–љ—Л–Љ —А–µ–љ—В–≥–µ–љ–Њ–≥—А–∞—Д–Є–Є –≥—А—Г–і–љ–Њ–є –Ї–ї–µ—В–Ї–Є – –Ј–∞—Б—В–Њ–є–љ—Л–µ —П–≤–ї–µ–љ–Є—П –≤ –ї–µ–≥–Ї–Є—Е, –і–≤—Г—Б—В–Њ—А–Њ–љ–љ–Є–є –≥–Є–і—А–Њ—В–Њ—А–∞–Ї—Б. –С–Њ–ї—М–љ–∞—П —Б—А–Њ—З–љ–Њ –≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–Є—А–Њ–≤–∞–љ–∞ –≤ –Ю–†–Ш–Ґ 3 –¶–µ–љ—В—А–∞–ї—М–љ–Њ–≥–Њ –≤–Њ–µ–љ–љ–Њ–≥–Њ –≥–Њ—Б–њ–Є—В–∞–ї—П –Є–Љ. –Р.–Т. –Т–Є—И–љ–µ–≤—Б–Ї–Њ–≥–Њ, –≥–і–µ –і–∞–љ–љ—Л—Е –≤ –њ–Њ–ї—М–Ј—Г –Њ—Б—В—А–Њ–≥–Њ –Є–љ—Д–∞—А–Ї—В–∞ –Љ–Є–Њ–Ї–∞—А–і–∞ –љ–µ –њ–Њ–ї—Г—З–µ–љ–Њ. –°–Њ—Б—В–Њ—П–љ–Є–µ –±—Л–ї–Њ —А–∞—Б—Ж–µ–љ–µ–љ–Њ, –Ї–∞–Ї –њ–Њ—Б—В–Є–љ—Д–∞—А–Ї—В–љ—Л–є –Ї–∞—А–і–Є–Њ—Б–Ї–ї–µ—А–Њ–Ј. –Э–∞ —Д–Њ–љ–µ –њ—А–Њ–≤–Њ–і–Є–Љ–Њ–є —В–µ—А–∞–њ–Є–Є –љ–Є—В—А–∞—В–∞–Љ–Є, –Є–љ–≥–Є–±–Є—В–Њ—А–∞–Љ–Є –Р–Я–§ –Є –Љ–Њ—З–µ–≥–Њ–љ–љ—Л–Љ–Є –њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є —Б–Њ—Б—В–Њ—П–љ–Є–µ —Г–ї—Г—З—И–Є–ї–Њ—Б—М (–Є—Б—З–µ–Ј–ї–Є –Њ—В–µ–Ї–Є –љ–∞ –љ–Њ–≥–∞—Е, —Г–Љ–µ–љ—М—И–Є–ї–∞—Б—М –Њ–і—Л—И–Ї–∞, –љ–∞ –Љ–Њ–Љ–µ–љ—В –≤—Л–њ–Є—Б–Ї–Є –ґ–Є–і–Ї–Њ—Б—В–Є –≤ –њ–ї–µ–≤—А–∞–ї—М–љ—Л—Е –њ–Њ–ї–Њ—Б—В—П—Е –љ–µ –Њ—В–Љ–µ—З–∞–ї–Њ—Б—М). –Ю—В –њ—А–µ–і–ї–Њ–ґ–µ–љ–љ–Њ–є –Ї–Њ—А–Њ–љ–∞—А–Њ–≥—А–∞—Д–Є–Є –Њ—В–Ї–∞–Ј–∞–ї–∞—Б—М. –Т–њ–Њ—Б–ї–µ–і—Б—В–≤–Є–Є –≤–љ–Њ–≤—М –љ–∞—А–∞—Б—В–∞—О—В —П–≤–ї–µ–љ–Є—П —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є, –њ–Њ—П–≤–Є–ї–∞—Б—М —В–µ–љ–і–µ–љ—Ж–Є—П –Ї –љ–Њ—А–Љ–Њ—В–Њ–љ–Є–Є. –Т –Є—О–љ–µ 2004 –≥–Њ–і–∞ –≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–Є—А—Г–µ—В—Б—П –≤ –Т–Ъ–Э–¶, –≥–і–µ –њ—А–Є –≠—Е–Њ–Ъ–У: –Ї–∞—А—В–Є–љ–∞ –≥–Є–њ–µ—А—В—А–Њ—Д–Є–Є –Љ–Є–Њ–Ї–∞—А–і–∞ –Ы–Ц (–Ґ–Ч–°––Ґ–Ь–Ц–Я – 14 –Љ–Љ), —А–∞—Б—И–Є—А–µ–љ–Є–µ –њ–Њ–ї–Њ—Б—В–Є –Ы–Я –і–Њ 4,2 —Б–Љ, –љ–∞—А—Г—И–µ–љ–Є–µ –і–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –Љ–Є–Њ–Ї–∞—А–і–∞ –Ы–Ц, –§–Т – 60%, –≤–њ–µ—А–≤—Л–µ –≤—Л—П–≤–ї–µ–љ—Л –њ—А–Є–Ј–љ–∞–Ї–Є –ї–µ–≥–Њ—З–љ–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є (–°–Ф–Ы–Р 45 –Љ–Љ —А—В.—Б—В.), –љ–∞—З–∞–ї—М–љ—Л–µ —П–≤–ї–µ–љ–Є—П —Б—В–µ–љ–Њ–Ј–∞ –ї–µ–≤–Њ–≥–Њ –Р–Т––Њ—В–≤–µ—А—Б—В–Є—П, –Љ–Є—В—А–∞–ї—М–љ–∞—П —А–µ–≥—Г—А–≥–Є—В–∞—Ж–Є—П 2 —Б—В. –Я—А–Њ–≤–µ–і–µ–љ–љ–∞—П –Ї–Њ—А–Њ–љ–∞—А–Њ–≥—А–∞—Д–Є—П –љ–µ –≤—Л—П–≤–Є–ї–∞ –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ —Б—В–µ–љ–Њ–Ј–Є—А–Њ–≤–∞–љ–Є—П –Ї–Њ—А–Њ–љ–∞—А–љ—Л—Е –∞—А—В–µ—А–Є–є. –°–Њ—Б—В–Њ—П–љ–Є–µ —А–∞—Б—Ж–µ–љ–Є–≤–∞–ї–Њ—Б—М, –Ї–∞–Ї —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ –≥–Є–њ–µ—А—В–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–µ—А–і—Ж–∞ —Г –±–Њ–ї—М–љ–Њ–є —Б –і–ї–Є—В–µ–ї—М–љ–Њ —Б—Г—Й–µ—Б—В–≤—Г—О—Й–µ–є –љ–µ–Ї–Њ—А—А–Є–≥–Є—А—Г–µ–Љ–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–µ–є, –Є–Ј–Љ–µ–љ–µ–љ–Є—П –љ–∞ –≠–Ъ–У –≤ –≤–Є–і–µ —А—Г–±—Ж–Њ–≤ –Є –љ–∞–ї–Є—З–Є–µ —Б–Є–љ–і—А–Њ–Љ–∞ —Б—В–µ–љ–Њ–Ї–∞—А–і–Є–Є —А–∞—Б—Ж–µ–љ–Є–≤–∞–ї–Є—Б—М, –Ї–∞–Ї –њ—А–Њ—П–≤–ї–µ–љ–Є—П –≥–Є–њ–µ—А—В—А–Њ—Д–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є–Є. –Э–∞–Ј–љ–∞—З–µ–љ–∞ —В–µ—А–∞–њ–Є—П –±–Є—Б–Њ–њ—А–Њ–ї–Њ–ї–Њ–Љ, —Н–љ–∞–ї–∞–њ—А–Є–ї–Њ–Љ, –≥–Є–і—А–Њ—Е–ї–Њ—А—В–Є–∞–Ј–Є–і–Њ–Љ. –£—А–Њ–≤–µ–љ—М –Р–Ф –љ–Њ—А–Љ–∞–ї–Є–Ј–Њ–≤–∞–ї—Б—П, –љ–Њ —Б–Њ—Е—А–∞–љ—П–ї–Є—Б—М –Њ–і—Л—И–Ї–∞ –Є —Б–ї–∞–±–Њ—Б—В—М. –£—Е—Г–і—И–µ–љ–Є–µ —Б–Њ—Б—В–Њ—П–љ–Є—П —Б —Д–µ–≤—А–∞–ї—П 2005 –≥–Њ–і–∞. –Т–љ–Њ–≤—М –љ–∞—А–∞—Б—В–∞–µ—В –Њ–і—Л—И–Ї–∞, —З—В–Њ –њ–Њ—В—А–µ–±–Њ–≤–∞–ї–Њ —Г—Б–Є–ї–µ–љ–Є—П –Љ–Њ—З–µ–≥–Њ–љ–љ–Њ–є —В–µ—А–∞–њ–Є–Є, –њ–Њ—П–≤–Є–ї–∞—Б—М —Б–Ї–ї–Њ–љ–љ–Њ—Б—В—М –Ї –≥–Є–њ–Њ—В–Њ–љ–Є–Є. –Т –∞–њ—А–µ–ї–µ 2005 –≤–Њ–Ј–љ–Є–Ї–ї–Є —Б—Г—Е–Њ—Б—В—М –Є –љ–µ–њ—А–Є—П—В–љ—Л–є –≤–Ї—Г—Б –≤–Њ —А—В—Г, –њ—А–Њ–њ–∞–ї –∞–њ–њ–µ—В–Є—В, –њ–Њ—П–≤–Є–ї–Є—Б—М —В—П–љ—Г—Й–Є–µ –±–Њ–ї–Є –≤ —Н–њ–Є–≥–∞—Б—В—А–Є–Є, –Њ–Ј–љ–Њ–±—Л –њ–Њ—Б–ї–µ –њ—А–Є–µ–Љ–∞ –њ–Є—Й–Є, –±–Њ–ї—М–љ–∞—П —Б—В–∞–ї–∞ —В–µ—А—П—В—М –≤ –≤–µ—Б–µ. –Я—А–Є –≠–У–Ф–° –њ–∞—В–Њ–ї–Њ–≥–Є–Є –љ–µ –≤—Л—П–≤–ї–µ–љ–Њ, –њ—А–Є –£–Ч–Ш –±—А—О—И–љ–Њ–є –њ–Њ–ї–Њ—Б—В–Є – –њ—А–Є–Ј–љ–∞–Ї–Є –і–Є—Д—Д—Г–Ј–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –њ–Њ–і–ґ–µ–ї—Г–і–Њ—З–љ–Њ–є –ґ–µ–ї–µ–Ј—Л, –і–≤—Г—Б—В–Њ—А–Њ–љ–љ–Є–є –≥–Є–і—А–Њ—В–Њ—А–∞–Ї—Б. –Т –Є—О–љ–µ –њ–Њ—П–≤–ї—П–µ—В—Б—П —Б—Г—Е–Њ–є –љ–∞–і—Б–∞–і–љ—Л–є –Ї–∞—И–µ–ї—М, –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—В –Њ–і—Л—И–Ї–∞ –Є —Б–ї–∞–±–Њ—Б—В—М. –Т –Њ–Ї—В—П–±—А–µ –±–Њ–ї—М–љ–∞—П –≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–Є—А—Г–µ—В—Б—П –≤ —Б—В–∞—Ж–Є–Њ–љ–∞—А, –Ї —Н—В–Њ–Љ—Г –Љ–Њ–Љ–µ–љ—В—Г –њ–Њ—В–µ—А—П –≤–µ—Б–∞ —Б–Њ—Б—В–∞–≤–Є–ї–∞ 10 –Ї–≥, —З—В–Њ –Ј–∞—Б—В–∞–≤–Є–ї–Њ –њ—А–Њ–≤–µ—Б—В–Є –Њ–љ–Ї–Њ–њ–Њ–Є—Б–Ї (–Ъ–Ґ –±—А—О—И–љ–Њ–є –њ–Њ–ї–Њ—Б—В–Є, –Ъ–Ґ –Љ–∞–ї–Њ–≥–Њ —В–∞–Ј–∞, –£–Ч–Ш –Љ–Њ–ї–Њ—З–љ—Л—Е –ґ–µ–ї–µ–Ј, —Й–Є—В–Њ–≤–Є–і–љ–Њ–є –ґ–µ–ї–µ–Ј—Л, –≥–µ–љ–Є—В–∞–ї–Є–є, –≠–У–Ф–°, –Ї–Њ–ї–Њ–љ–Њ—Б–Ї–Њ–њ–Є—П), –љ–µ –≤—Л—П–≤–Є–≤—И–Є–є –Њ–љ–Ї–Њ–њ–∞—В–Њ–ї–Њ–≥–Є–Є. –Я—А–Є –Ъ–Ґ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –Њ—А–≥–∞–љ–Њ–≤ –≥—А—Г–і–љ–Њ–є –Ї–ї–µ—В–Ї–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ: –њ—А–∞–≤–Њ—Б—В–Њ—А–Њ–љ–љ–Є–є –њ–ї–µ–≤—А–∞–ї—М–љ—Л–є –≤—Л–њ–Њ—В, –Ј–Њ–љ–∞ —Г–њ–ї–Њ—В–љ–µ–љ–Є—П –ї–µ–≥–Њ—З–љ–Њ–є —В–Ї–∞–љ–Є –і–Є–∞–Љ–µ—В—А–Њ–Љ 10 –Љ–Љ –≤ –• —Б–µ–≥–Љ–µ–љ—В–µ –њ—А–∞–≤–Њ–≥–Њ –ї–µ–≥–Ї–Њ–≥–Њ, –Њ–±–Њ–≥–∞—Й–µ–љ–Є–µ —Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ —А–Є—Б—Г–љ–Ї–∞ –Ј–∞ —Б—З–µ—В —Г—В–Њ–ї—Й–µ–љ–Є—П –Љ–µ–ґ–і–Њ–ї—М–Ї–Њ–≤—Л—Е –њ–µ—А–µ–≥–Њ—А–Њ–і–Њ–Ї. –≠—В–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Є–љ—В–µ—А–њ—А–µ—В–Є—А–Њ–≤–∞–љ—Л –Ї–∞–Ї –њ—А–∞–≤–Њ—Б—В–Њ—А–Њ–љ–љ—П—П –љ–Є–ґ–љ–µ–і–Њ–ї–µ–≤–∞—П –њ–љ–µ–≤–Љ–Њ–љ–Є—П, –њ–ї–µ–≤—А–∞–ї—М–љ—Л–є –≤—Л–њ–Њ—В —Б–њ—А–∞–≤–∞, –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–є –Њ—В–µ–Ї –Є–ї–Є –ї–Є–Љ—Д–Њ–≥–µ–љ–љ—Л–є –Ї–∞—А—Ж–Є–љ–Њ–Љ–∞—В–Њ–Ј. –Я—А–Њ–≤–µ–і–µ–љ–∞ –њ–ї–µ–≤—А–∞–ї—М–љ–∞—П –њ—Г–љ–Ї—Ж–Є—П, –њ—А–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ —В—А–∞–љ—Б—Б—Г–і–∞—В, –∞—В–Є–њ–Є—З–љ—Л—Е –Ї–ї–µ—В–Њ–Ї –љ–µ –љ–∞–є–і–µ–љ–Њ. –Т –±–Є–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Є—Е (—Н–ї–µ–Ї—В—А–Њ—Д–Њ—А–µ–Ј –±–µ–ї–Ї–Њ–≤—Л—Е —Д—А–∞–Ї—Ж–Є–є –љ–µ –њ—А–Њ–≤–Њ–і–Є–ї—Б—П) –Є –Є–Љ–Љ—Г–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –∞–љ–∞–ї–Є–Ј–∞—Е –Ї—А–Њ–≤–Є – –±–µ–Ј –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —Б–і–≤–Є–≥–Њ–≤, –Ј–∞ –Є—Б–Ї–ї—О—З–µ–љ–Є–µ–Љ 10––Ї—А–∞—В–љ–Њ–≥–Њ –њ–Њ–≤—Л—И–µ–љ–Є—П —Г—А–Њ–≤–љ—П –Њ–љ–Ї–Њ–≥–µ–љ–∞ —П–Є—З–љ–Є–Ї–Њ–≤ (–°–Р 125). –Я—А–Њ–≤–Њ–і–Є–ї–Є—Б—М –∞–љ—В–Є–±–∞–Ї—В–µ—А–Є–∞–ї—М–љ–∞—П, –≥–Є–њ–Њ—В–µ–љ–Ј–Є–≤–љ–∞—П (–њ–µ—А–Є–љ–і–Њ–њ—А–Є–ї, –љ–µ–±–Є–≤–Њ–ї–Њ–ї) –Є –Љ–Њ—З–µ–≥–Њ–љ–љ–∞—П (–≥–Є–і—А–Њ—Е–ї–Њ—А—В–Є–∞–Ј–Є–і 100–Љ–≥) —В–µ—А–∞–њ–Є—П, —Б –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–Љ —Г–ї—Г—З—И–µ–љ–Є–µ–Љ –≤—Л–њ–Є—Б–∞–љ–∞ –і–Њ–Љ–Њ–є. –°–Њ—Б—В–Њ—П–љ–Є–µ –±–Њ–ї—М–љ–Њ–є –Љ–µ–і–ї–µ–љ–љ–Њ —Г—Е—Г–і—И–∞–µ—В—Б—П, –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г–µ—В –Њ–і—Л—И–Ї–∞, –љ–∞–Ї–∞–њ–ї–Є–≤–∞–µ—В—Б—П –ґ–Є–і–Ї–Њ—Б—В—М –≤ –њ–ї–µ–≤—А–∞–ї—М–љ—Л—Е –њ–Њ–ї–Њ—Б—В—П—Е, –њ—А–Є—Б–Њ–µ–і–Є–љ—П—О—В—Б—П –љ–Њ—З–љ—Л–µ –Њ—А—В–Њ–њ–љ–Њ—Н –±–µ–Ј –њ–Њ–≤—Л—И–µ–љ–Є—П —Г—А–Њ–≤–љ—П –Р–Ф, —З—В–Њ –Ј–∞—Б—В–∞–≤–ї—П–µ—В –њ—А–Њ–і–Њ–ї–ґ–∞—В—М –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–є –њ–Њ–Є—Б–Ї. –Я—А–Є –њ–Њ–≤—В–Њ—А–љ—Л—Е –Ъ–Ґ––Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –ї–µ–≥–Ї–Є—Е –≤—Л—П–≤–ї—П–µ—В—Б—П —Г—Б–Є–ї–µ–љ–Є–µ –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —А–Є—Б—Г–љ–Ї–∞, –Ї–Њ—В–Њ—А–Њ–µ —А–∞—Б—Ж–µ–љ–Є–≤–∞–µ—В—Б—П, –Ї–∞–Ї –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–є –Є –љ–∞—З–Є–љ–∞—О—Й–Є–є—Б—П –∞–ї—М–≤–µ–Њ–ї—П—А–љ—Л–є –Њ—В–µ–Ї –ї–µ–≥–Ї–Є—Е.
–С–Њ–ї—М–љ–∞—П –њ–Њ—Б—В—Г–њ–∞–µ—В –≤ –њ—Г–ї—М–Љ–Њ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Њ—В–і–µ–ї–µ–љ–Є–µ –Ї–ї–Є–љ–Є–Ї–Є –≥–Њ—Б–њ–Є—В–∞–ї—М–љ–Њ–є —В–µ—А–∞–њ–Є–Є –Ь–Ь–Р –Є–Љ–µ–љ–Є –Ш.–Ь. –°–µ—З–µ–љ–Њ–≤–∞. –Я—А–Є –њ–Њ—Б—В—Г–њ–ї–µ–љ–Є–Є: —Б–Њ—Б—В–Њ—П–љ–Є–µ —Б—А–µ–і–љ–µ–є —В—П–ґ–µ—Б—В–Є. –Р—Б—В–µ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ —В–µ–ї–Њ—Б–ї–Њ–ґ–µ–љ–Є—П, –њ–Њ–љ–Є–ґ–µ–љ–љ–Њ–≥–Њ –њ–Є—В–∞–љ–Є—П. –Т–µ—Б 50 –Ї–≥, —А–Њ—Б—В – 162 —Б–Љ. –Ш–Ь–Ґ – 19. –Ъ–Њ–ґ–љ—Л–µ –њ–Њ–Ї—А–Њ–≤—Л –±–ї–µ–і–љ—Л–µ, —Б—Г—Е–Є–µ. –¶–Є–∞–љ–Њ–Ј –≥—Г–±. –Я–∞–ї—М–њ–Є—А—Г—О—В—Б—П –њ–Њ–і–Љ—Л—И–µ—З–љ—Л–µ –ї–Є–Љ—Д–Њ—Г–Ј–ї—Л. –Т–∞—А–Є–Ї–Њ–Ј–љ–Њ–µ —А–∞—Б—И–Є—А–µ–љ–Є–µ –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В–љ—Л—Е –≤–µ–љ –љ–Є–ґ–љ–Є—Е –Ї–Њ–љ–µ—З–љ–Њ—Б—В–µ–є. –У–Њ–ї–µ–љ–Є –њ–∞—Б—В–Њ–Ј–љ—Л, –±–Њ–ї—М—И–µ – –њ—А–∞–≤–∞—П (–Ј–∞ —Б—З–µ—В –≤–µ–љ–Њ–Ј–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є). –Э–Њ—Б–Њ–≤–Њ–µ –і—Л—Е–∞–љ–Є–µ —Б–≤–Њ–±–Њ–і–љ–Њ–µ. –І–Ф–Ф – 20 –≤ 1 –Љ–Є–љ. –≤ –њ–Њ–Ї–Њ–µ. –У—А—Г–і–љ–∞—П –Ї–ї–µ—В–Ї–∞ –Њ–±—Л—З–љ–Њ–є —Д–Њ—А–Љ—Л, –њ—А–∞–≤–∞—П –њ–Њ–ї–Њ–≤–Є–љ–∞ –Њ—В—Б—В–∞–µ—В –≤ –∞–Ї—В–µ –і—Л—Е–∞–љ–Є—П. –Т –љ–Є–ґ–љ–Є—Е –Њ—В–і–µ–ї–∞—Е –Њ–±–Њ–Є—Е –ї–µ–≥–Ї–Є—Е –њ—А–Є –њ–µ—А–Ї—Г—Б—Б–Є–Є –Њ–њ—А–µ–і–µ–ї—П–ї–Њ—Б—М –њ—А–Є—В—Г–њ–ї–µ–љ–Є–µ –і–Њ —Г—А–Њ–≤–љ—П 5 —А–µ–±—А–∞. –Ф—Л—Е–∞–љ–Є–µ –љ–∞–і –ї–µ–≥–Ї–Є–Љ–Є –≤–µ–Ј–Є–Ї—Г–ї—П—А–љ–Њ–µ —Б –ґ–µ—Б—В–Ї–Є–Љ –Њ—В—В–µ–љ–Ї–Њ–Љ, —Б–њ—А–∞–≤–∞ –≤ –±–∞–Ј–∞–ї—М–љ—Л—Е –Њ—В–і–µ–ї–∞—Е –і—Л—Е–∞–љ–Є–µ –љ–µ –њ—А–Њ–≤–Њ–і–Є—В—Б—П, —Б–ї–µ–≤–∞ – –Њ—Б–ї–∞–±–ї–µ–љ–љ–Њ–µ. –Ю–±–ї–∞—Б—В—М —Б–µ—А–і—Ж–∞ –љ–µ –Є–Ј–Љ–µ–љ–µ–љ–∞. –У—А–∞–љ–Є—Ж—Л –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ–є —В—Г–њ–Њ—Б—В–Є —Б–µ—А–і—Ж–∞ —А–∞—Б—И–Є—А–µ–љ—Л –≤–ї–µ–≤–Њ. –Ґ–Њ–љ—Л —Б–µ—А–і—Ж–∞ –њ—А–Є–≥–ї—Г—И–µ–љ—Л, —А–Є—В–Љ–Є—З–љ—Л–µ, –І–°–°– 80 —Г–і. –≤ –Љ–Є–љ. –Р–Ї—Ж–µ–љ—В 2 —В–Њ–љ–∞ –љ–∞ –∞–Њ—А—В–µ –Є –ї–µ–≥–Њ—З–љ–Њ–є –∞—А—В–µ—А–Є–Є, –≤ –Њ–±–ї–∞—Б—В–Є –≤–µ—А—Е—Г—И–Ї–Є –≤—Л—Б–ї—Г—И–Є–≤–∞–µ—В—Б—П —Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–Є–є —И—Г–Љ. –Р–Ф – 90/60 –Љ–Љ —А—В.—Б—В. –Ц–Є–≤–Њ—В –њ—А–Є –њ–∞–ї—М–њ–∞—Ж–Є–Є –Љ—П–≥–Ї–Є–є, —З—Г–≤—Б—В–≤–Є—В–µ–ї–µ–љ –≤ —Н–њ–Є–≥–∞—Б—В—А–Є–Є, –≤ –Њ—Б—В–∞–ї—М–љ—Л—Е –Њ—В–і–µ–ї–∞—Е –±–µ–Ј–±–Њ–ї–µ–Ј–љ–µ–љ–љ—Л–є. –Я–µ—З–µ–љ—М –Њ–њ—Г—Й–µ–љ–∞, –Ї—А–∞–є —А–Њ–≤–љ—Л–є, –±–µ–Ј–±–Њ–ї–µ–Ј–љ–µ–љ–љ—Л–є. –°–µ–ї–µ–Ј–µ–љ–Ї–∞ –љ–µ —Г–≤–µ–ї–Є—З–µ–љ–∞. –Я–Њ—З–Ї–Є –љ–µ –њ–∞–ї—М–њ–Є—А—Г—О—В—Б—П. –°–Є–Љ–њ—В–Њ–Љ –њ–Њ–Ї–Њ–ї–∞—З–Є–≤–∞–љ–Є—П – –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–є. –Ф–Є–Ј—Г—А–Є–Є –љ–µ—В. –У—А—Г–±–Њ–є –љ–µ–≤—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–Є –љ–µ—В.
–Р–љ–∞–ї–Є–Ј—Л –Ї—А–Њ–≤–Є: –°–Ю–≠ – 3 –Љ–Љ/—З, –ї–µ–є–Ї–Њ—Ж–Є—В—Л – 11000 , –љ–µ–є—В—А–Њ—Д–Є–ї—Л – 74%, —Н–Њ–Ј–Є–љ–Њ—Д–Є–ї—Л – 4 %, –Љ–Њ–љ–Њ—Ж–Є—В—Л – 2%, –ї–Є–Љ—Д–Њ—Ж–Є—В—Л – 16%, –±–∞–Ј–Њ—Д–Є–ї—Л – 1 %. –У–µ–Љ–Њ–≥–ї–Њ–±–Є–љ –145 –≥ /–ї, —Н—А–Є—В—А–Њ—Ж–Є—В—Л – 4,7 –Љ–ї–љ, —В—А–Њ–Љ–±–Њ—Ж–Є—В—Л – 443 000, –¶–Я–0,92. –Њ–±—Й. –±–µ–ї–Њ–Ї – 7 (6–8) –≥/–і–ї, –∞–ї—М–±—Г–Љ–Є–љ– 4,1 –≥/–і–ї, –Ї—А–µ–∞—В–Є–љ–Є–љ – 1,1 –Љ–≥/–і–ї. –Я—А–Є —Н–ї–µ–Ї—В—А–Њ—Д–Њ—А–µ—В–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –±–µ–ї–Ї–Њ–≤—Л—Е —Д—А–∞–Ї—Ж–Є–є –≤—Л—П–≤–ї–µ–љ –Ь––≥—А–∞–і–Є–µ–љ—В –≤ –Ј–Њ–љ–µ –≥–∞–Љ–Љ–∞––≥–ї–Њ–±—Г–ї–Є–љ–Њ–≤, –њ—А–Є –Є–Љ–Љ—Г–љ–Њ—Е–Є–Љ–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –±–µ–ї–Ї–Њ–≤ —Б—Л–≤–Њ—А–Њ—В–Ї–Є –Ї—А–Њ–≤–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ–∞ –њ–∞—А–∞–њ—А–Њ—В–µ–Є–љ–µ–Љ–Є—П G, —Б–Њ—Б—В–∞–≤–ї—П–≤—И–∞—П 12,7% –Њ—В –Њ–±—Й–µ–≥–Њ –±–µ–ї–Ї–∞ —Б—Л–≤–Њ—А–Њ—В–Ї–Є –Ї—А–Њ–≤–Є, –Є–ї–Є 9,1 –≥/–ї, —Г—А–Њ–≤–µ–љ—М –±–µ—В–∞–2––Љ–Є–Ї—А–Њ–≥–ї–Њ–±—Г–ї–Є–љ–∞ –Є –°–†–С – –≤ –љ–Њ—А–Љ–µ.
–Я–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –Њ–љ–Ї–Њ–Љ–∞—А–Ї–µ—А–∞ –°–Р 125 –≤ 3 —А–∞–Ј–∞.
–Т –Њ–±—Й–µ–Љ –∞–љ–∞–ї–Є–Ј–µ –Љ–Њ—З–Є —Б–ї–µ–і–Њ–≤–∞—П –њ—А–Њ—В–µ–Є–љ—Г—А–Є—П, –≤ –Њ—Б—В–∞–ї—М–љ–Њ–Љ –±–µ–Ј –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Њ—В–Ї–ї–Њ–љ–µ–љ–Є–є. –≠–ї–µ–Ї—В—А–Њ—Д–Њ—А–µ—В–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –±–µ–ї–Ї–Њ–≤ –Љ–Њ—З–Є –љ–µ –≤—Л—П–≤–Є–ї–Њ –±–µ–ї–Ї–∞ –С–µ–љ—Б––Ф–ґ–Њ–љ—Б–∞ (–≤ —В–Њ–Љ —З–Є—Б–ї–µ –њ—А–Є –Є–Љ–Љ—Г–љ–Њ—Д–Є–Ї—Б–∞—Ж–Є–Є —Б –∞–љ—В–Є—Б—Л–≤–Њ—А–Њ—В–Ї–Њ–є –Ї —Б–≤–Њ–±–Њ–і–љ—Л–Љ –ї–µ–≥–Ї–Є–Љ —Ж–µ–њ—П–Љ).
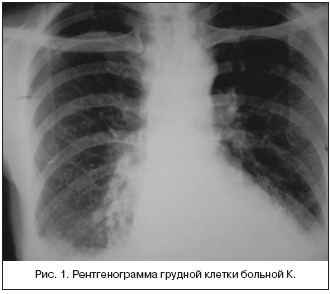
–Я—А–Є —А–µ–љ—В–≥–µ–љ–Њ–≥—А–∞—Д–Є–Є –Њ—А–≥–∞–љ–Њ–≤ –≥—А—Г–і–љ–Њ–є –Ї–ї–µ—В–Ї–Є –Њ–њ—А–µ–і–µ–ї—П–ї–∞—Б—М –ґ–Є–і–Ї–Њ—Б—В—М –≤ –њ–ї–µ–≤—А–∞–ї—М–љ—Л—Е –њ–Њ–ї–Њ—Б—В—П—Е —Б–њ—А–∞–≤–∞ –і–Њ 6 —А–µ–±—А–∞, —Б–ї–µ–≤–∞ – 6 –Љ–µ–ґ—А–µ–±–µ—А—М–µ. –Т –љ–Є–ґ–љ–Є—Е –і–Њ–ї—П—Е –Њ–±–Њ–Є—Е –ї–µ–≥–Ї–Є—Е –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П –і–Є—Д—Д—Г–Ј–љ–∞—П –і–µ—Д–Њ—А–Љ–∞—Ж–Є—П –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —А–Є—Б—Г–љ–Ї–∞, –Є–Љ–µ—О—Й–∞—П —Б–µ—В—З–∞—В–Њ–—П—З–µ–Є—Б—В–Њ–µ —Б—В—А–Њ–µ–љ–Є–µ, —Б–Њ—Б—Г–і–Є—Б—В—Л–є —А–Є—Б—Г–љ–Њ–Ї –≤ —Н—В–Є—Е –Ј–Њ–љ–∞—Е –љ–µ –њ—А–Њ—Б–ї–µ–ґ–Є–≤–∞–ї—Б—П. –Т–µ—А—Е—Г—И–Ї–Є –ї–µ–≥–Ї–Є—Е –љ–µ –Є–Ј–Љ–µ–љ–µ–љ—Л. –Ъ–Њ—А–љ–Є –ї–µ–≥–Ї–Є—Е –љ–µ —А–∞—Б—И–Є—А–µ–љ—Л, —Б—В—А—Г–Ї—В—Г—А–љ—Л, —И–Є—А–Є–љ–∞ –ї–µ–≥–Њ—З–љ–Њ–є –∞—А—В–µ—А–Є–Є –љ–∞ —Г—А–Њ–≤–љ–µ –њ—А–∞–≤–Њ–≥–Њ –ї–µ–≥–Ї–Њ–≥–Њ 1,3 —Б–Љ (–љ–Њ—А–Љ–∞). –°–µ—А–і—Ж–µ –≥–Њ—А–Є–Ј–Њ–љ—В–∞–ї—М–љ–Њ —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–Њ. –Р–Њ—А—В–∞ —А–∞—Б—И–Є—А–µ–љ–∞, —Г–і–ї–Є–љ–µ–љ–∞ (—А–Є—Б.1).
–Я—А–Є –Ъ–Ґ –ї–µ–≥–Ї–Є—Е – –≤ –њ–ї–µ–≤—А–∞–ї—М–љ—Л—Е –њ–Њ–ї–Њ—Б—В—П—Е –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П –ґ–Є–і–Ї–Њ—Б—В—М, –±–Њ–ї—М—И–µ –≤ –њ—А–∞–≤–Њ–є. –Ы–µ–≥–Њ—З–љ—Л–є —А–Є—Б—Г–љ–Њ–Ї —Г—Б–Є–ї–µ–љ –≤–Њ –≤—Б–µ—Е –Њ—В–і–µ–ї–∞—Е –Ј–∞ —Б—З–µ—В —Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–∞. –Ю—В–Љ–µ—З–∞–ї–Њ—Б—М –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –ї–Є–Љ—Д–Њ—Г–Ј–ї–Њ–≤ —Б—А–µ–і–Њ—Б—В–µ–љ–Є—П.
–Я—А–Є –≠—Е–Њ–Ъ–У –≤—Л—П–≤–ї—П–ї–∞—Б—М –Ї–Њ–љ—Ж–µ–љ—В—А–Є—З–µ—Б–Ї–∞—П –≥–Є–њ–µ—А—В—А–Њ—Д–Є—П –Љ–Є–Њ–Ї–∞—А–і–∞ –Ы–Ц (–Ґ–Ь–Ц–Я––Ґ–Ч–°–14 –Љ–Љ) —Б –і–Є–ї–∞—В–∞—Ж–Є–µ–є –њ–Њ–ї–Њ—Б—В–Є –Ы–Я –і–Њ 4,8 —Б–Љ, –Ъ–Ф–†/–Ъ–°–† –Ы–Ц —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ 4,9/3,6 —Б–Љ, –§–Т – 52%, —А–∞–Ј–Љ–µ—А—Л –Я–Ц – 2,7 —Б–Љ, —Г–Љ–µ—А–µ–љ–љ–∞—П –≥–Є–њ–µ—А—В—А–Њ—Д–Є—П —Б—В–µ–љ–Ї–Є –Я–Ц – 0,52 —Б–Љ. –Э–µ–±–Њ–ї—М—И–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –≥–ї–Њ–±–∞–ї—М–љ–Њ–є —Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –Ы–Ц. –§–Є–±—А–Њ–Ј —Б—В–≤–Њ—А–Њ–Ї –Ь–Ъ, —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –≥—А–∞–і–Є–µ–љ—В–∞ –і–∞–≤–ї–µ–љ–Є—П –љ–∞ –Ь–Ъ –і–Њ 8 –Љ–Љ —А—В.—Б—В., –њ–ї–Њ—Й–∞–і—М —Б–µ—З–µ–љ–Є—П 3,7 —Б–Љ, –Њ–і–љ–∞–Ї–Њ –і–≤–Є–ґ–µ–љ–Є–µ —Б—В–≤–Њ—А–Њ–Ї –Ь–Ъ –≤ –њ—А–Њ—В–Є–≤–Њ—Д–∞–Ј–µ. –†–µ–≥—Г—А–≥–Є—В–∞—Ж–Є—П –Ь–Ъ 2–3 —Б—В–µ–њ–µ–љ–Є. –Ф–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –Ы–Ц –њ–Њ 2 —В–Є–њ—Г. –°—А–µ–і–љ–µ–µ –і–∞–≤–ї–µ–љ–Є–µ –≤ –Ы–Р 20 –Љ–Љ —А—В. —Б—В. (–њ—А–Є –љ–Њ—А–Љ–µ – –і–Њ 15 –Љ–Љ —А—В. —Б—В.). –Э–µ–±–Њ–ї—М—И–Њ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –ґ–Є–і–Ї–Њ—Б—В–Є –≤ –њ–Њ–ї–Њ—Б—В–Є –њ–µ—А–Є–Ї–∞—А–і–∞. –Ю–±—А–∞—Й–∞–ї –љ–∞ —Б–µ–±—П –≤–љ–Є–Љ–∞–љ–Є–µ —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–є –Њ–њ–∞–ї–µ—Б—Ж–Є—А—Г—О—Й–Є–є, —Г—Б–Є–ї–µ–љ–љ—Л–є —Н—Е–Њ—Б–Є–≥–љ–∞–ї –Њ—В —Б—В–µ–љ–Њ–Ї –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞. –Т–њ–µ—А–≤—Л–µ –≤—Л—Б–Ї–∞–Ј–∞–љ–Њ –њ—А–µ–і–њ–Њ–ї–Њ–ґ–µ–љ–Є–µ –Њ–± –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–µ —Б–µ—А–і—Ж–∞.
–§–Т–Ф – –Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–µ –≤–µ–љ—В–Є–ї—П—Ж–Є–Њ–љ–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П –њ–Њ —А–µ—Б—В—А–Є–Ї—В–Є–≤–љ–Њ–Љ—Г —В–Є–њ—Г (–Ц–Х–Ы–1,93 –ї (66%), –Ю–§–Т1–1,53 –ї (62%).
–С–Њ–ї—М–љ–Њ–є –±—Л–ї–∞ –≤—Л–њ–Њ–ї–љ–µ–љ–∞ –њ—Г–љ–Ї—Ж–Є—П –њ—А–∞–≤–Њ–є –њ–ї–µ–≤—А–∞–ї—М–љ–Њ–є –њ–Њ–ї–Њ—Б—В–Є, —Н–≤–∞–Ї—Г–Є—А–Њ–≤–∞–љ–Њ 500 –Љ–ї —Б–Њ–ї–Њ–Љ–µ–љ–љ–Њ––ґ–µ–ї—В–Њ–є –њ—А–Њ–Ј—А–∞—З–љ–Њ–є –ґ–Є–і–Ї–Њ—Б—В–Є. –Я—А–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Г–і–µ–ї—М–љ—Л–є –≤–µ—Б 1003, –±–µ–ї–Њ–Ї 5,05%, –≥–ї—О–Ї–Њ–Ј–∞ 150 –Љ–≥%, –њ—А–Њ–±–∞ –†–Є–≤–∞–ї—М—В–∞ – –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ–∞—П, –њ—А–Є —Ж–Є—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є – –ї–µ–є–Ї–Њ—Ж–Є—В–Њ–≤ – 5–10–15, –ї–Є–Љ—Д–Њ—Ж–Є—В–Њ–≤ – 75%, –љ–µ–є—В—А–Њ—Д–Є–ї–Њ–≤ – 25%, —Н—А–Є—В—А–Њ—Ж–Є—В–Њ–≤ – 40–60. –Р—В–Є–њ–Є—З–љ—Л–µ –Ї–ї–µ—В–Ї–Є –Є –С–Ъ –љ–µ –љ–∞–є–і–µ–љ—Л. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П–Љ–Є –Љ–µ—В–Њ–і–Њ–Љ –Я–¶–† –Є –ї—О–Љ–Є–љ–Є—Б—Ж–µ–љ—В–љ–Њ–є –Љ–Є–Ї—А–Њ—Б–Ї–Њ–њ–Є–Є –Љ–Є–Ї–Њ–±–∞–Ї—В–µ—А–Є–Є —В—Г–±–µ—А–Ї—Г–ї–µ–Ј–∞ –љ–µ –≤—Л—П–≤–ї–µ–љ—Л.
–Т —Б—В–µ—А–љ–∞–ї—М–љ–Њ–Љ –њ—Г–љ–Ї—В–∞—В–µ –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ –њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –Ї–ї–µ—В–Њ–Ї (–і–Њ 1,8%).
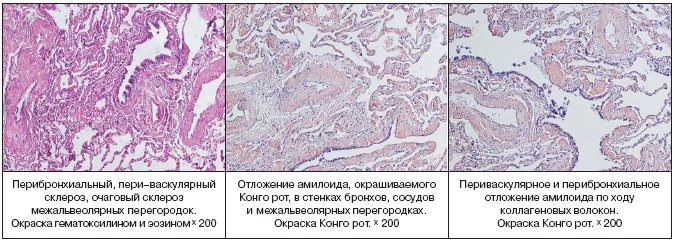
–Ю–±—Б—Г–ґ–і–∞–ї–Є—Б—М –≤–µ—А–Њ—П—В–љ—Л–µ –і–Є–∞–≥–љ–Њ–Ј—Л: —Б–Є–љ–і—А–Њ–Љ –Ь–µ–є–≥—Б–∞, –Љ–µ–Ј–Њ—В–µ–ї–Є–Њ–Љ–∞ –њ–ї–µ–≤—А—Л, –ї–Є–Љ—Д–Њ–љ–≥–∞–Є—В –ї–µ–≥–Ї–Є—Е, —В—Г–±–µ—А–Ї—Г–ї–µ–Ј. –Т—Л–њ–Њ–ї–љ–µ–љ–љ–Њ–µ –£–Ч–Ш –Љ–∞–ї–Њ–≥–Њ —В–∞–Ј–∞ –љ–µ –≤—Л—П–≤–Є–ї–Њ –њ–∞—В–Њ–ї–Њ–≥–Є–Є —Б–Њ —Б—В–Њ—А–Њ–љ—Л –њ—А–∞–≤–Њ–≥–Њ —П–Є—З–љ–Є–Ї–∞, –±—Л–ї–Є –њ–µ—А–µ—Б–Љ–Њ—В—А–µ–љ—Л –±–ї–Њ–Ї–Є –±–Є–Њ–њ—В–∞—В–Њ–≤ —Г–і–∞–ї–µ–љ–љ—Л—Е –≤ 2001 –≥–Њ–і—Г –Љ–∞—В–Ї–Є –Є –ї–µ–≤–Њ–≥–Њ —П–Є—З–љ–Є–Ї–∞ – –і–∞–љ–љ—Л—Е, –њ–Њ–і—В–≤–µ—А–ґ–і–∞—О—Й–Є—Е –љ–µ–Њ–њ–ї–∞—Б—В–Є—З–µ—Б–Ї–Є–є –њ—А–Њ—Ж–µ—Б—Б, –љ–µ –њ–Њ–ї—Г—З–µ–љ–Њ. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –і–Є–∞–≥–љ–Њ–Ј —Б–Є–љ–і—А–Њ–Љ–∞ –Ь–µ–є–≥—Б–∞ —Б—В–∞–ї –Ї—А–∞–є–љ–µ —Б–Њ–Љ–љ–Є—В–µ–ї—М–љ—Л–Љ. –Ю—В—Б—Г—В—Б—В–≤–Є–µ –Љ–Є–Ї–Њ–±–∞–Ї—В–µ—А–Є–є —В—Г–±–µ—А–Ї—Г–ї–µ–Ј–∞ –њ—А–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –њ–ї–µ–≤—А–∞–ї—М–љ–Њ–є –ґ–Є–і–Ї–Њ—Б—В–Є, —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Ї–Є—Е –Є –Њ—З–∞–≥–Њ–≤ –≤–љ–µ –ї–µ–≥–Њ—З–љ–Њ–≥–Њ —В—Г–±–µ—А–Ї—Г–ї–µ–Ј–∞ –њ–Њ–Ј–≤–Њ–ї—П–ї–Њ –Є—Б–Ї–ї—О—З–∞—В—М —В—Г–±–µ—А–Ї—Г–ї–µ–Ј–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Ф–ї—П —Г—В–Њ—З–љ–µ–љ–Є—П –і–Є–∞–≥–љ–Њ–Ј–∞ –≤—Л–њ–Њ–ї–љ–µ–љ–∞ –≤–Є–і–µ–Њ—В–Њ—А–∞–Ї–Њ—Б–Ї–Њ–њ–Є—П —Б –∞—В–Є–њ–Є—З–љ–Њ–є —А–µ–Ј–µ–Ї—Ж–Є–µ–є –≤–µ—А—Е–љ–µ–є –і–Њ–ї–Є –њ—А–∞–≤–Њ–≥–Њ –ї–µ–≥–Ї–Њ–≥–Њ. –Т —Е–Њ–і–µ –Њ–њ–µ—А–∞—Ж–Є–Є –∞—Б–њ–Є—А–Є—А–Њ–≤–∞–љ–Њ 1,5 –ї–Є—В—А–∞ –њ—А–Њ–Ј—А–∞—З–љ–Њ–є –ґ–µ–ї—В–Њ–≥–Њ —Ж–≤–µ—В–∞ –ґ–Є–і–Ї–Њ—Б—В–Є. –Я–∞—А–Є–µ—В–∞–ї—М–љ–∞—П –Є –≤–Є—Б—Ж–µ—А–∞–ї—М–љ–∞—П –њ–ї–µ–≤—А–∞ –љ–µ —Г—В–Њ–ї—Й–µ–љ–∞, —Б–Њ—Б—Г–і—Л –Є–љ—К–µ—Ж–Є—А–Њ–≤–∞–љ—Л. –Ю—В–Љ–µ—З–∞–ї–Њ—Б—М –њ–Њ–ї–љ–Њ–Ї—А–Њ–≤–Є–µ –ї–µ–≥–Ї–Њ–≥–Њ. –Т 1 —Б–µ–≥–Љ–µ–љ—В–µ —Г—З–∞—Б—В–Њ–Ї —А—Г–±—Ж–Њ–≤–Њ–≥–Њ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –ї–µ–≥–Њ—З–љ–Њ–є —В–Ї–∞–љ–Є —Б —Г—В–Њ–ї—Й–µ–љ–љ–Њ–є –±–µ–ї–µ—Б–Њ–≤–∞—В–Њ–є –њ–ї–µ–≤—А–Њ–є. –Я—А–Њ–≤–µ–і–µ–љ –Љ–µ—Е–∞–љ–Є—З–µ—Б–Ї–Є–є –Є —Е–Є–Љ–Є—З–µ—Б–Ї–Є–є –њ–ї–µ–≤—А–Њ–і–µ–Ј —Б –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ 1% —А–∞—Б—В–≤–Њ—А–∞ –є–Њ–і–Њ–њ–Є—А–Њ–љ–∞. –Я—А–Є –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є – –≤ –њ—А–µ–њ–∞—А–∞—В–∞—Е —В–Ї–∞–љ—М –ї–µ–≥–Ї–Њ–≥–Њ —Б —Д–Є–±—А–Њ–Ј–Њ–Љ –њ–ї–µ–≤—А—Л. –Т —Д–Є–±—А–Њ–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–є –њ–ї–µ–≤—А–µ –Њ—З–∞–≥–Њ–≤—Л–µ –ї–Є–Љ—Д–Њ–≥–Є—Б—В–Є–Њ—Ж–Є—В–∞—А–љ—Л–µ –Є–љ—Д–Є–ї—М—В—А–∞—В—Л, —Б–Ї–ї–µ—А–Њ–Ј —Б–Њ—Б—Г–і–Њ–≤ –Є –Љ–µ—Б—В–∞–Љ–Є —П–≤–ї–µ–љ–Є—П –ї–Є–Љ—Д–Њ—Б—В–∞–Ј–∞, –Ї—А–Њ–≤–Њ–Є–Ј–ї–Є—П–љ–Є—П. –Т —Б—Г–±–њ–ї–µ–≤—А–∞–ї—М–љ—Л—Е –Њ—В–і–µ–ї–∞—Е –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–є —Д–Є–±—А–Њ–Ј –Є –≥–Є–∞–ї–Є–љ–Њ–Ј, –≤ –њ—А–Њ—Б–≤–µ—В–µ –Њ—В–і–µ–ї—М–љ—Л—Е –∞–ї—М–≤–µ–Њ–ї –Ї—А—Г–њ–љ—Л–µ –Љ–∞–Ї—А–Њ—Д–∞–≥–Є —Б –±—Г—А—Л–Љ –њ–Є–≥–Љ–µ–љ—В–Њ–Љ –≤ —Ж–Є—В–Њ–њ–ї–∞–Ј–Љ–µ. –Я—А–Є –Њ–Ї—А–∞—Б–Ї–µ –Ъ–Њ–љ–≥–Њ–—А–Њ—В –Є —В–Є–Њ—Д–ї–∞–≤–Є–љ–Њ–Љ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ –Њ—В–ї–Њ–ґ–µ–љ–Є–µ –∞–Љ–Є–ї–Њ–Є–і–∞ –≤ —Б—В–µ–љ–Ї–∞—Е —Б–Њ—Б—Г–і–Њ–≤, –ї–µ–≥–Њ—З–љ–Њ–Љ –Є–љ—В–µ—А—Б—В–Є—Ж–Є–Є –Є –≤ –њ–ї–µ–≤—А–µ. –Я—А–Њ–≤–µ–і–µ–љ–љ–Њ–µ —В–Є–њ–Є—А–Њ–≤–∞–љ–Є–µ –њ–Њ–і—В–≤–µ—А–і–Є–ї–Њ –љ–∞–ї–Є—З–Є–µ AL––∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –У–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ —Б—Л–≤–Њ—А–Њ—В–Ї–Є –Ї—А–Њ–≤–Є –љ–µ –≤—Л—П–≤–Є–ї–Њ –Љ—Г—В–∞—Ж–Є–є –≤ –≥–µ–љ–µ —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–∞.
–Я–Њ—Б–ї–µ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –і–Є–∞–≥–љ–Њ–Ј–∞ –±–Њ–ї—М–љ–∞—П –Њ—В–Ї–∞–Ј–∞–ї–∞—Б—М –Њ—В –ї–µ—З–µ–љ–Є—П. –Т –і–∞–ї—М–љ–µ–є—И–µ–Љ –±–Њ–ї—М–љ–∞—П –≥–Њ—Б–њ–Є—В–∞–ї–Є–Ј–Є—А—Г–µ—В—Б—П –≤ –ї–µ—З–µ–±–љ–Њ–µ —Г—З—А–µ–ґ–і–µ–љ–Є–µ –≥. –Ь–Њ—Б–Ї–≤—Л, –≥–і–µ, —Б –µ–µ —Б–ї–Њ–≤, –±—Л–ї –њ—А–Њ–≤–µ–і–µ–љ –Ї—Г—А—Б —В–µ—А–∞–њ–Є–Є –Љ–µ–ї—Д–∞–ї–∞–љ–Њ–Љ –Є –њ—А–µ–і–љ–Є–Ј–Њ–ї–Њ–љ–Њ–Љ –≤ —Й–∞–і—П—Й–Є—Е –і–Њ–Ј–∞—Е. –Т—В–Њ—А–Њ–є –Ї—Г—А—Б –Є–Ј––Ј–∞ –њ–ї–Њ—Е–Њ–є –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В–Є –ї–µ—З–µ–љ–Є—П –љ–µ –њ—А–Њ–≤–Њ–і–Є–ї—Б—П. –І–µ—А–µ–Ј 4 –Љ–µ—Б. –±–Њ–ї—М–љ–∞—П —Б–Ї–Њ–љ—З–∞–ї–∞—Б—М –њ—А–Є –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є—Е —П–≤–ї–µ–љ–Є—П—Е —Б–µ—А–і–µ—З–љ–Њ–—Б–Њ—Б—Г–і–Є—Б—В–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є.
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Г –њ–∞—Ж–Є–µ–љ—В–Ї–Є –і–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞–љ –РL––∞–Љ–Є–ї–Њ–Є–і–Њ–Ј —Б –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –ї–µ–≥–Ї–Є—Е –Є –њ–ї–µ–≤—А—Л (–і–Є—Д—Д—Г–Ј–љ–Њ––Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ—Л–є —В–Є–њ), —Б–µ—А–і—Ж–∞ (–љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М –Ї—А–Њ–≤–Њ–Њ–±—А–∞—Й–µ–љ–Є—П II –Р —Б—В–∞–і–Є—П —Б –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –і–Є–∞—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ —А–µ—Б—В—А–Є–Ї—В–Є–≤–љ–Њ–Љ—Г —В–Є–њ—Г, –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ —Б —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ–Љ —Б—В–µ–љ–Њ–Ј–∞ –ї–µ–≤–Њ–≥–Њ –РV––Њ—В–≤–µ—А—Б—В–Є—П), –≤–µ–≥–µ—В–∞—В–Є–≤–љ–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л (–Њ—А—В–Њ—Б—В–∞—В–Є—З–µ—Б–Ї–∞—П –≥–Є–њ–Њ—В–µ–љ–Ј–Є—П, –Њ—Е—А–Є–њ–ї–Њ—Б—В—М –≥–Њ–ї–Њ—Б–∞), —Б–Њ—Б—Г–і–Њ–≤.
–Я—А–Є —А–µ—В—А–Њ—Б–њ–µ–Ї—В–Є–≤–љ–Њ–Љ –∞–љ–∞–ї–Є–Ј–µ –∞–љ–∞–Љ–љ–µ–Ј–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–∞—Ж–Є–µ–љ—В–Ї–Є –Љ–Њ–ґ–љ–Њ —Г—В–≤–µ—А–ґ–і–∞—В—М, —З—В–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –і–µ–±—О—В–Є—А–Њ–≤–∞–ї–Њ –≤ 1998 –≥–Њ–і—Г —Б –Ї–Њ–ґ–љ—Л—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є – –њ–Њ—П–≤–ї–µ–љ–Є–µ –њ–µ—В–µ—Е–Є–∞–ї—М–љ—Л—Е –≤—Л—Б—Л–њ–∞–љ–Є–є –≤ –њ–∞—А–∞–Њ—А–±–Є—В–∞–ї—М–љ—Л—Е –Ј–Њ–љ–∞—Е. –Я–Њ—П–≤–ї–µ–љ–Є–µ –Њ—Б–Є–њ–ї–Њ—Б—В–Є –≥–Њ–ї–Њ—Б–∞ –≤ 2000 –≥–Њ–і—Г —П–≤–ї—П–ї–Њ—Б—М –њ–µ—А–≤—Л–Љ –њ—А–Є–Ј–љ–∞–Ї–Њ–Љ –≤–µ–≥–µ—В–∞—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є, —З—В–Њ –Њ—И–Є–±–Њ—З–љ–Њ —А–∞—Б—Ж–µ–љ–Є–≤–∞–ї–Њ—Б—М –±–Њ–ї—М–љ–Њ–є –Є –≤—А–∞—З–∞–Љ–Є, –Ї–∞–Ї –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–µ –њ–Њ—Б–ї–µ –њ–µ—А–µ–љ–µ—Б–µ–љ–љ–Њ–≥–Њ –≥—А–Є–њ–њ–∞. –Т –њ–Њ—Б–ї–µ–і—Г—О—Й–µ–Љ —Д–Њ—А–Љ–Є—А—Г–µ—В—Б—П –≥–Є–њ–µ—А—В—А–Њ—Д–Є—З–µ—Б–Ї–∞—П –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є—П, —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–≤—И–∞—П—Б—П –≤ –њ—А–Њ–≥—А–∞–Љ–Љ–µ –љ–µ–Ї–Њ—А—А–Є–≥–Є—А—Г–µ–Љ–Њ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–Є. –Ю—З–µ–љ—М –Є–љ—В–µ—А–µ—Б–љ—Л–є —Д–∞–Ї—В – —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ —Б—В–µ–љ–Њ–Ј–∞ –њ–Њ–љ–∞—З–∞–ї—Г –Њ–±—Б—Г–ґ–і–∞–ї–Њ—Б—М –≤ –њ—А–Њ–≥—А–∞–Љ–Љ–µ —А–µ–≤–Љ–∞—В–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П; –њ–Њ—Б–ї–µ —В–Њ–≥–Њ –Ї–∞–Ї —А–µ–≤–Љ–∞—В–Є–Ј–Љ –±—Л–ї –Њ—В–≤–µ—А–≥–љ—Г—В, –Њ—Б—В–∞–љ–Њ–≤–Є–ї–Є—Б—М –љ–∞ –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ—В–Є—З–µ—Б–Ї–Њ–Љ –≥–µ–љ–µ–Ј–µ, –љ–µ—Б–Љ–Њ—В—А—П –љ–∞ —В–Њ, —З—В–Њ –Ї–Њ—А–Њ–љ–∞—А–љ—Л–µ —Б–Њ—Б—Г–і—Л (–њ–Њ –і–∞–љ–љ—Л–Љ –Ї–Њ—А–Њ–љ–∞—А–Њ–≥—А–∞—Д–Є–Є) –±—Л–ї–Є –Є–љ—В–∞–Ї—В–љ—Л. –Ґ–µ–њ–µ—А—М —Г–ґ–µ —Б —Г–≤–µ—А–µ–љ–љ–Њ—Б—В—М—О –Љ–Њ–ґ–љ–Њ —Г—В–≤–µ—А–ґ–і–∞—В—М, —З—В–Њ –њ–Њ—А–∞–ґ–µ–љ–Є–µ —Д–Є–±—А–Њ–Ј–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞ –ї–µ–≤–Њ–≥–Њ –РV––Њ—В–≤–µ—А—Б—В–Є—П –Є —Б—В–≤–Њ—А–Њ–Ї –Ь–Ъ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ –Њ—В–ї–Њ–ґ–µ–љ–Є–µ–Љ –≤ –љ–Є—Е –∞–Љ–Є–ї–Њ–Є–і–∞. –Я–µ—А–≤—Л–µ –њ—А–Є–Ј–љ–∞–Ї–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П –ї–µ–≥–Ї–Є—Е –њ–Њ—П–≤–Є–ї–Є—Б—М –≤ 2003 –≥–Њ–і—Г; —В–Њ–≥–і–∞ –Њ–љ–Є –љ–µ–≤–µ—А–љ–Њ –Є–љ—В–µ—А–њ—А–µ—В–Є—А–Њ–≤–∞–ї–Є—Б—М, –Ї–∞–Ї –њ—А–Њ—П–≤–ї–µ–љ–Є—П —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є. –Э–∞–Ї–Њ–њ–ї–µ–љ–Є–µ —В—А–∞–љ—Б—Б—Г–і–∞—В–∞ –≤ –њ–ї–µ–≤—А–∞–ї—М–љ—Л—Е –њ–Њ–ї–Њ—Б—В—П—Е –њ—А–Є —Б–Њ—Е—А–∞–љ–љ–Њ–є —Б–Њ–Ї—А–∞—В–Є—В–µ–ї—М–љ–Њ–є —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є –Љ–Є–Њ–Ї–∞—А–і–∞ –Є –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л—Е –њ—А–Є–Ј–љ–∞–Ї–∞—Е –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –Ї—А–Њ–≤–Њ–Њ–±—А–∞—Й–µ–љ–Є—П (–Њ–≥—А–∞–љ–Є—З–Є–≤–∞—О—Й–µ–є—Б—П —В–Њ–ї—М–Ї–Њ –Љ–∞–ї—Л–Љ –Ї—А—Г–≥–Њ–Љ) —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤–Њ–≤–∞–ї–Є –Њ –њ–Њ—А–∞–ґ–µ–љ–Є–Є –њ–ї–µ–≤—А—Л –Є –њ–Њ—В–µ—А–Є –µ–µ –±–∞—А—М–µ—А–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є. –Р —А–∞–Ј–≤–Є—В–Є–µ —Б—В–Њ–є–Ї–Њ–є –≥–Є–њ–Њ—В–µ–љ–Ј–Є–Є —Г –±–Њ–ї—М–љ–Њ–є, –і–ї–Є—В–µ–ї—М–љ–Њ —Б—В—А–∞–і–∞—О—Й–µ–є –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–Њ–љ–Є–µ–є, –±—Л–ї–Њ —Б–≤—П–Ј–∞–љ–Њ —Б –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –≤–µ–≥–µ—В–∞—В–Є–≤–љ–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л, –∞ –љ–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–є —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О.
–Э–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –≤–Њ–Ј–љ–Є–Ї–љ–Њ–≤–µ–љ–Є–µ –њ–µ—В–µ—Е–Є–∞–ї—М–љ—Л—Е –≤—Л—Б—Л–њ–∞–љ–Є–є –≤ –њ–∞—А–∞–Њ—А–±–Є—В–∞–ї—М–љ—Л—Е –Њ–±–ї–∞—Б—В—П—Е —П–≤–ї—П–µ—В—Б—П —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–Љ –і–ї—П AL––∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ —Б–Є–Љ–њ—В–Њ–Љ–Њ–Љ –Є —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–µ—В—Б—П, –Ї–∞–Ї –њ–∞—В–Њ–≥–љ–Њ–Љ–Њ–љ–Є—З–љ—Л–є –Є –њ—А–Њ–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–є –њ—А–Є–Ј–љ–∞–Ї –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–≥–Њ —В–µ—З–µ–љ–Є—П –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –љ–Њ, –≤–Њ–њ—А–µ–Ї–Є –Њ–±—Й–µ–њ—А–Є–љ—П—В–Њ–Љ—Г –Љ–љ–µ–љ–Є—О, –≤ –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–љ–Њ–Љ –љ–∞–Љ–Є —Б–ї—Г—З–∞–µ –Њ—В –љ–∞—З–∞–ї–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –і–Њ —Д–Є–љ–∞–ї–∞ –њ—А–Њ—И–ї–Њ 8 (!) –ї–µ—В. –Т–Њ–Ј–Љ–Њ–ґ–љ–Њ, –њ—А–∞–≤–Є–ї—М–љ–∞—П –Њ—Ж–µ–љ–Ї–∞ —Н—В–Њ–≥–Њ —Б–Є–Љ–њ—В–Њ–Љ–∞ –≤ –љ–∞—З–∞–ї–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –њ–Њ–Ј–≤–Њ–ї–Є–ї–∞ –±—Л —Г—Б—В–∞–љ–Њ–≤–Є—В—М –і–Є–∞–≥–љ–Њ–Ј –љ–∞ –±–Њ–ї–µ–µ —А–∞–љ–љ–µ–Љ —Н—В–∞–њ–µ –Є –і–∞–ї–∞ –±–Њ–ї—М—И–µ —И–∞–љ—Б–Њ–≤ –і–ї—П –ї–µ—З–µ–љ–Є—П –Є –њ—А–Њ–і–ї–µ–љ–Є—П –ґ–Є–Ј–љ–Є –њ–∞—Ж–Є–µ–љ—В–Ї–Є.
–°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В—М—О –і–∞–љ–љ–Њ–≥–Њ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–ї—Г—З–∞—П —П–≤–ї—П–µ—В—Б—П –Њ—В—Б—Г—В—Б—В–≤–Є–µ –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–Њ—З–µ–Ї –і–∞–ґ–µ –≤ —А–∞–Ј–≤–µ—А–љ—Г—В–Њ–є —Б—В–∞–і–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, —З—В–Њ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –Ј–∞—В—А—Г–і–љ–Є–ї–Њ –њ–Њ–і—Е–Њ–і—Л –Ї –і–Є–∞–≥–љ–Њ–Ј—Г.
–Ґ–Њ–ї—М–Ї–Њ –Њ–±—К–µ–і–Є–љ–µ–љ–Є–µ —А–∞–Ј–љ–Њ–Њ–±—А–∞–Ј–љ—Л—Е, –љ–∞ –њ–µ—А–≤—Л–є –≤–Ј–≥–ї—П–і, —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Њ –њ–Њ–і–Њ–є—В–Є –Ї –і–Є–∞–≥–љ–Њ–Ј—Г –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, –Ї–Њ—В–Њ—А—Л–є –±—Л–ї –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є.
–Т –љ–∞—И–µ–Љ —Б–Њ–Њ–±—Й–µ–љ–Є–Є –Љ—Л –±—Л —Е–Њ—В–µ–ї–Є –њ—А–Є–≤–ї–µ—З—М –≤–љ–Є–Љ–∞–љ–Є–µ –Ї –њ–∞—В–Њ–ї–Њ–≥–Є–Є, –і–Њ–≤–Њ–ї—М–љ–Њ —А–µ–і–Ї–Њ –≤—Б—В—А–µ—З–∞—О—Й–µ–є—Б—П –Ї–∞–Ї –≤ –њ—Г–ї—М–Љ–Њ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є, —В–∞–Ї –Є –Њ–±—Й–µ—В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Њ–є –њ—А–∞–Ї—В–Є–Ї–µ. –Ґ—А—Г–і–љ–Њ—Б—В—М –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є, –Ї–∞–Ї —Г–ґ–µ –Њ—В–Љ–µ—З–∞–ї–Њ—Б—М, —Б–≤—П–Ј–∞–љ–∞ —Б –Љ–љ–Њ–≥–Њ–Њ–±—А–∞–Ј–Є–µ–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є–є –њ–µ—А–≤–Є—З–љ–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Я—А–Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–Є –Њ–±—Й–µ–њ—А–Є–љ—П—В—Л—Е —Б—В–∞–љ–і–∞—А—В–Њ–≤ –і–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –њ–Њ–Є—Б–Ї–∞ —В–µ—А—П–µ—В—Б—П –њ—А–Є–љ—Ж–Є–њ –Є–љ–і–Є–≤–Є–і—Г–∞–ї—М–љ–Њ–≥–Њ –њ–Њ–і—Е–Њ–і–∞ –Ї –њ–∞—Ж–Є–µ–љ—В—Г, —З—В–Њ –Ј–∞—В—А—Г–і–љ—П–µ—В –Њ–њ—А–µ–і–µ–ї–µ–љ–Є–µ –Є—Б—В–Є–љ–љ–Њ–≥–Њ –і–Є–∞–≥–љ–Њ–Ј–∞. –Ш–Ј—Г—З–∞—П —А–∞–Ј—А–∞–±–Њ—В–∞–љ–љ—Л–µ –Є —А–µ–Ї–Њ–Љ–µ–љ–і–Њ–≤–∞–љ–љ—Л–µ —Б—Е–µ–Љ—Л –і–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ–Њ–≥–Њ –њ–Њ–Є—Б–Ї–∞ –њ—А–Є –њ–ї–µ–≤—А–∞–ї—М–љ—Л—Е –≤—Л–њ–Њ—В–∞—Е –Њ—В–µ—З–µ—Б—В–≤–µ–љ–љ—Л—Е –∞–≤—В–Њ—А–Њ–≤, –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Ј–∞–Љ–µ—В–Є—В—М, —З—В–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –≤ —Н—В–Њ—В –Ї—А—Г–≥ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –љ–µ –≤—Е–Њ–і–Є—В. –Ь–µ–ґ–і—Г —В–µ–Љ –≤ —Б—В–∞—В—М—П—Е –Ј–∞—А—Г–±–µ–ґ–љ—Л—Е –∞–≤—В–Њ—А–Њ–≤ –љ–∞ —Н—В—Г —В–µ–Љ—Г –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј –≤–Ї–ї—О—З–µ–љ –≤ –њ–Њ–Є—Б–Ї –њ—А–Є—З–Є–љ –њ–ї–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –≤—Л–њ–Њ—В–∞ [11].
–Ы–Є—В–µ—А–∞—В—Г—А–∞
1. Gertz M.A., Kyle R.A., Thibodeau S.N. Familial amyloidosis: a study of 52 North American– born patients examined during a 30–year period. Mayo Clin Proc 1992; 67:428–440.
2. Simms R.W, Prout M.N, Cohen A.S. The epidemiology of AL and AA amyloidosis. Baillieres Clin Rheumatol 1994;8:627–634.
3. Kyle R.A, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995;32:45–59.
4. Koyama J, Ray–Sequin PA, Davidoff R, et al. Usefulness of pulsed tissue Doppler imaging for evaluating systolic and diastolic left ventricular function in patients with AL (prima¬ry) amyloidosis. Am J Cardiol 2002; 89:1067–1071.
5. –Т–Є–љ–Њ–≥—А–∞–і–Њ–≤–∞ –Ю.–Ь.. –Я–µ—А–≤–Є—З–љ—Л–є –Є –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –≤–∞—А–Є–∞–љ—В—Л –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞. –Ь., «–Ь–µ–і–Є—Ж–Є–љ–∞», 1980.
6. –Ь–Њ–Є—Б–µ–µ–≤ –°.–Т. –Ш–љ—Д–Є–ї—М—В—А–∞—В–Є–≤–љ—Л–µ –њ–Њ—А–∞–ґ–µ–љ–Є—П –Љ–Є–Њ–Ї–∞—А–і–∞. –†–µ—Б—В—А–Є–Ї—В–Є–≤–љ–∞—П –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є—П. –Ь.,–§–∞—А–Љ–∞ –Я—А–µ—Б—Б,1998.
7. Berk J.L., O’Regan A., Skinner M. Pulmonary and tracheobronchial amyloidosis. Semin Respirat Crit Care Med 2002; 23:155–165.
8. Berk J.L., Keane J., Seldin D.C., et al. Persistent pleural effusions in primary systemic amyloidosis. Chest 2003; 124;969–977.
9. Mathews V, Vasudevan AR, Nair S, et al. Primary systemicamyloidosis: pleural involvement with exudative pleural effu¬sion. J Assoc Physicians India 1996; 44:345–346
10. –Ъ–Њ–Ј–ї–Њ–≤—Б–Ї–∞—П –Ы.–Т., –Т–∞—А—И–∞–≤—Б–Ї–Є–є –Т.–Р., –І–µ–≥–∞–µ–≤–∞ –Ґ.–Т. –Є –і—А. –Р–Љ–Є–ї–Њ–Є–і–Њ–Ј —Б–Њ–≤—А–µ–Љ–µ–љ–љ—Л–є –≤–Ј–≥–ї—П–і –љ–∞ –њ—А–Њ–±–ї–µ–Љ—Г. //–Я—А–∞–Ї—В–Є—З–µ—Б–Ї–∞—П –љ–µ—Д—А–Њ–ї–Њ–≥–Є—П. — 1998. — 2:24–26.
11. Pleural effusion: Article bu Fredrick M Abrahamian, DO Jan.11.2005.



